Force Hemato lance chaque année deux appels d’offres auprès des différents groupes coopérateurs afin de soutenir des projets de recherche innovants :
- Appel d’offre 1 : «Etudes de biologie reliées à des protocoles de recherche clinique, des études translationnelles ou des essais précoces» dotés chacun de 50 000€
- Appel d’offre 2 : «Etudes portant sur l’épidémiologie, la pharmaco-économie, l’éducation thérapeutique, les sciences humaines, la qualité de vie et l’accompagnement des patients»dotés de 25 000€
Prix Brigitte MERAND :
- Ce prix récompense chaque année les travaux d’un chercheur âgé de moins de 45 ans
- Dotation : 8 000€
Prix de thèse :
- Ce prix récompense chaque année 2 thésards.
- Dotation : 2 500€ par projet
Projets de Recherche
ÉTUDES DE BIOLOGIE RELIÉES À DES PROTOCOLES DE RECHERCHE CLINIQUE, DES ÉTUDES TRANSLATIONNELLES OU DES ESSAIS PRÉCOCES

Vahid ASNAFI
CARACTÉRISATION DE MARQUEURS BIOLOGIQUES DE RÉSISTANCE AU TRAITEMENT DANS LES LEUCÉMIES AIGUËS LYMPHOBLASTIQUES T
Les leucémies aiguës lymphoblastiques T (LAL-T) sont des cancers hématologiques rares qui affectent principalement les adolescents et les jeunes adultes. Contrairement aux leucémies de phénotype B qui ont bénéficié de l’essor de thérapies innovantes comme les CAR T-cells ou encore les anticorps bispécifiques, le pronostic des LAL-T est profondément altéré en cas de chimiorésistance, étant donné le peu d’alternatives thérapeutiques disponibles. Il est donc primordial de rechercher des marqueurs de résistance à la chimiothérapie afin d’identifier précocement ces patients et d’améliorer leur prise en charge et leur pronostic. Des études préliminaires dans le laboratoire ont permis d’identifier des gènes potentiellement impliqués dans la résistance à la chimiothérapie des LAL-T, en comparant les profils de régulation épigénétique de patients sensibles à la chimiothérapie à des patients résistants à la chimiothérapie. Nous souhaitons développer un test biologique qui permettrait d’identifier dès le diagnostic les patients à haut risque de chimiorésistance afin de pouvoir leur proposer rapidement des alternatives thérapeutiques. Nous souhaitons également étudier comment les gènes identifiés pourraient être impliqués dans cette résistance au traitement et ouvrir la voie au développement de potentielles thérapies ciblées.

Gregory LAZARIAN
ÉVALUATION DE LA MALADIE RÉSIDUELLE MOLÉCULAIRE PAR SÉQUENÇAGE NGS DU RÉARRANGEMENT DES GÈNES DES IMMUNOGLOBULINES SUR L’ADN TUMORAL CIRCULANT PLASMATIQUE DANS LA LEUCÉMIE LYMPHOÏDE CHRONIQUE ET LE SYNDROME DE RICHTER
Dans la leucémie lymphoïde chronique, l’évaluation de la maladie résiduelle (ou détection de cellules tumorales résiduelles minimes par des techniques très sensibles) a pris une place importante pour évaluer la réponse au traitement et prédire l’évolution clinique des patients.
Les techniques actuellement utilisées pour évaluer la maladie résiduelle sont basées sur la détection directement de cellules tumorales dans le sang ou dans la moelle osseuse mais elles ne prennent pas en compte la présence possible de cellules persistantes dans le tissu ganglionnaire, qui pourraient être à l’origine de récidive de la maladie. Pour mieux apprécier la maladie résiduelle en considérant toutes les localisations possibles de la maladie dans l’organisme, nous souhaitons mettre en place une nouvelle technique qui permet de détecter dans le sang l’ADN tumoral circulant issus de la mort des cellules leucémiques, quelles que soit leurs localisations sanguine, ganglionnaire ou médullaire.
Notre projet consistera à analyser des échantillons de plasma collectés et congelés dans le cadre d’un essai clinique chez des patients avant et après traitement pour valider la faisabilité de la technique pour la détection de l’ADN tumoral circulant comme marqueur de maladie résiduelle.

David SIBON
CARACTÉRISATION GÉNOMIQUE ET TRANSCRIPTOMIQUE DU LYMPHOME ANAPLASIQUE À GRANDES CELLULES ALK-POSITIF : ÉTUDE BIO-ALK-0BS, ANCILLAIRE DE L’ÉTUDE PROSPECTIVE ALK-OBS DU LYSA
Le lymphome anaplasique à grandes cellules ALK-positif est un lymphome rare survenant chez des patients jeunes majoritairement entre 10 et 40 ans. Environ 50 nouveaux cas (10 enfants et 40 adultes) sont diagnostiqués chaque année en France. Ce lymphome est lié à la survenue du dérèglement d’un gène particulier appelé ALK.
Après chimiothérapie, la plupart des patients sont guéris. Cependant, pour les patients qui rechutent, le pronostic est sombre et les décès fréquents. Il est donc particulièrement important d’identifier dès le diagnostic initial les patients à haut risque de rechute, afin de pouvoir leur proposer, à terme, un traitement adapté. Pour cela, notre projet a pour objectif de décrire la carte d’identité moléculaire de ce lymphome en analysant l’ADN et l’ARN des cellules tumorales, afin de trouver des caractéristiques permettant de prédire le risque de rechute. A cette fin, nous analyserons les cellules tumorales de 40 patients ayant participé à l’étude prospective ALK-OBS du groupe coopérateur international LYSA (the Lymphoma Study Association), entre 2018 et 2021.
Les résultats attendus sont particulièrement importants, car la carte d’identité moléculaire de ce lymphome n’a jamais été étudiée de façon précise. Ces résultats pourront potentiellement modifier le traitement des patients et améliorer leurs chances de guérison.

Camille ROUSSEL
SPHÉROMARK: MARQUEURS DE SÉVÉRITÉ DANS LA SPHÉROCYTOSE HÉRÉDITAIRE
La sphérocytose héréditaire est une maladie génétique responsable d’anomalies au niveau des constituants de la membrane (enveloppe) des globules rouges. Ces anomalies rendent les globules rouges plus sphériques et plus rigides et entrainent leur destruction prématurée par la rate. En effet, la rate est un filtre pour les globules rouges, constituant un véritable « contrôle qualité » : les globules rouges sains et déformables passent le filtre tandis que les globules rouges trop vieux, présentant des anomalies ou pas assez déformables pour circuler, sont retenus et éliminés.
Dans la sphérocytose héréditaire, les globules rouges vivent donc moins longtemps ce qui entraine une anémie et l’augmentation du volume de la rate (splénomégalie). Lorsque l’anémie est trop profonde, on doit retirer la rate (c’est ce qu’on appelle une splénectomie) pour éviter la destruction des globules rouges, mais cela expose à des complications (notamment des infections) à long terme.
À ce jour, il n’existe pas de marqueur de gravité de la maladie permettant de prédire quel patient devra avoir une splénectomie. Nos équipes ont développés 2 outils innovants permettant d’une part, à l’aide de microbilles métalliques, de mimer au laboratoire le rôle de la rate dans la filtration des globules rouges (technique de microsphiltration) et d’autre part d’étudier simultanément et à haut débit les modifications de formes et les altérations des constituants de la membrane des globules rouges sphérocytaires (par la technique d’imagerie en flux).
L’objectif de notre projet est de trouver, à l’aide de ces 2 techniques innovantes, des marqueurs de gravité de la maladie, qui pourraient guider les médecins dans la prise en charge de la maladie, et en particulier dans la décision de pratiquer une splénectomie.

Augustin BOUDRY
ÉTUDE DE LA MALADIE RÉSIDUELLE DANS LES LEUCÉMIES AIGUËS MYÉLOÏDES PAR SÉQUENÇAGE HAUT DÉBIT AU SEIN DE LA COHORTE ALFA-1200
La leucémie aiguë myéloblastique (LAM) est une forme de cancer du sang résultant de l’accumulation de mutations cancéreuses. Le pronostic de cette maladie demeure sombre car les rechutes sont fréquentes après une réponse initiale au traitement. La persistance de cellules leucémiques en faible nombre après traitement (nommée maladie résiduelle) est un facteur de mauvais pronostic, augmentant le risque de rechute et diminuant la survie des patients. Les cellules leucémiques persistantes peuvent être décelées grâce à leurs mutations cancéreuses. Les techniques recommandées actuellement pour évaluer la maladie résiduelle permettent de rechercher une seule mutation à la fois et sont lourdes à mettre en place au laboratoire. Le séquençage à haut débit est une alternative intéressante car il permet de détecter toutes les mutations associées aux LAM en une seule analyse. Actuellement, cette technique n’est pas employée pour estimer la maladie résiduelle car il est difficile de distinguer les anomalies génétiques « vraies » qui sont effectivement présentes dans les cellules cancéreuses du patient, des anomalies génétiques « fausses » résultant d’erreurs liées au séquençage à haut débit (artefacts de séquençage).
Le projet de recherche vise à étudier la maladie résiduelle chez 157 patients âgés de plus de 60 ans atteints de LAM (cohorte ALFA-1200). Un programme bio-informatique sera développé pour analyser les résultats de séquençage de façon homogène, automatisée et reproductible. L’objectif final est d’évaluer l’intérêt de la maladie résiduelle pour la sélection du traitement, notamment pour le recours à une transplantation de cellules souches hématopoïétiques.
Cette étude permettra de détecter plus précisément la maladie restante dans la leucémie aiguë myéloïde (LAM) et d’identifier les patients à risque de rechute, améliorant ainsi le traitement et la survie des patients atteints de LAM.

Alexandre GUY
ÉTUDE ANCILLAIRE À L’ESSAI CLINIQUE AVAJAK : ÉTUDE PROSPECTIVE D’ÉVALUATION DE LA VALEUR PRONOSTIQUE DES MARQUEURS DE NÉTOSE POUR PRÉDIRE LA THROMBOSE AU COURS DES NÉOPLASIES MYÉLOPROLIFÉRATIVES AVEC MUTATION JAK2V617F (AVATARE)
Les néoplasies myélo-prolifératives (NMP) sont des maladies sanguines caractérisées par une production excessive d’une ou plusieurs lignées sanguines. Le risque principal de ces maladies est le développement d’une thrombose (« caillot ») au niveau d’une veine et ou d’une artère. Des progrès importants ont été faits dans la compréhension des mécanismes de ces thromboses mais, à ce jour, cette complication est toujours responsable d’une morbi-mortalité importante chez les patients avec NMP. Notre équipe étudie depuis plusieurs années un type de globules blancs, les polynucléaires neutrophiles, qui sont capables d’expulser de l’ADN lorsqu’ils sont activés : des filets extracellulaires ou (en anglais) neutrophil extracellular traps (NETs). De manière intéressante, il a été montré que les NETs participaient aux mécanismes de thrombose. Dans cette étude, qui va s’appuyer sur une étude internationale de grande ampleur (étude AVAJAK), nous souhaitons regarder à différentes périodes la formation de NETs chez des patients avec NMP à haut risque de faire une thrombose. Les patients seront soit traités par aspirine, soit par un nouvel anticoagulant oral. Nous serons ainsi en mesure d’étudier de nouveaux et nombreux biomarqueurs dans une population de patients avec NMP de grande taille et analyser plusieurs paramètres : l’association entre la formation de NETs et la survenue de thrombose, l’évolution du taux de NETs entre l’entrée dans l’étude et le suivi ultérieur (12 mois), l’effet des traitements (aspirine, anticoagulant) sur les taux de NETs.
L’accomplissement de cette étude permettra ainsi de mieux comprendre la thrombose au cours des NMP, d’identifier de nouveaux biomarqueurs de thrombose et ainsi de pouvoir à l’avenir développer de nouvelles pistes de traitement pour ces complications graves.
ÉTUDES PORTANT SUR L’ÉPIDÉMIOLOGIE, LA PHARMACO-ÉCONOMIE, L’ÉDUCATION THÉRAPEUTIQUE, LES SCIENCES HUMAINES, LA QUALITÉ DE VIE ET L’ACCOMPAGNEMENT DES PATIENTS

Maël HEIBLIG
ÉTABLISSEMENT D’UN JEU DE CRITÈRES PRIORITAIRES, STANDARDISÉS DANS LE SYNDROME DE VEXAS ET CONSTRUCTION D’UN SCORE SYMPTOMATIQUE PERSONNEL DU VEXAS (VPSS) ÉVALUANT LA CHARGE SYMPTOMATIQUE DE PATIENTS ATTEINTS DU SYNDROME DE VEXAS
Le syndrome VEXAS (pour Vacuoles in myeloid progenitors, E1 ubiquitin ligase, X-linked, Autoinflammatory manifestations and Somatic) est la conséquence de l’expansion de cellules souches de la moelle osseuse lié à une anomalie génétique acquise affectant le gène UBA1. Les patients atteints du syndrome de VEXAS présentent une variété de manifestations inflammatoires associées à de nombreuses complications (thrombose, infections) et l’apparition progressive de cytopénies tel que l’anémie. Le syndrome de VEXAS se caractérise par une profonde altération de l’état général et de la qualité de vie des patients se traduisant par un pronostic défavorable. Sur le plan thérapeutique, il n’existe à ce jour aucun traitement de référence. Des résultats préliminaires suggèrent que des médicaments utilisés dans d’autre maladie du sang pourraient permettre de contrôler les symptomes induits par la maladie. Cependant, ces résultats ne reposent pas sur des études cliniques controlées. Il existe donc un besoin urgent de mettre en place des essais cliniques afin d’évaluer l’intérêt de ces stratégies, afin d’améliorer la qualité de vie et la survie globale des patients. Cependant, une des limites majeures à la mise en place d’essais cliniques évaluant l’efficacité thérapeutique est l’absence de critères d’évaluation objectifs de réponses, spécifiquement dédiés à cette pathologie. Dans ce contexte, nous souhaitons mettre en place une étude permettant de déterminer un jeu de critères jugés importants par les patients et cliniciens et l’élaboration d’un score symptomatique par le biais d’instruments de mesure des résultats liés aux patients (Patient Reported Outcomes (PROs)). A terme et une fois validés, ces critères et ce score pourraient être utilisés pour évaluer l’efficacité de différents traitements en cours de développement dans le syndrome de VEXAS.

Gurvan HERMANGE
APPLICATION EN LIGNE D’AIDE À LA DÉCISION CLINIQUE POUR LE TRAITEMENT À L’IFN ALPHA DE PATIENTS ATTEINTS DE NMP
L’Interféron alpha (IFNα) a ouvert une porte d’espoir dans le traitement des certains cancers du sang appelés Néoplasmes Myéloprolifératifs (NMP). Bien que prometteur, ce traitement n’offre pas les mêmes résultats pour tous les patients. Certains répondent remarquablement bien, tandis que d’autres montrent une réaction plus modérée ou même négligeable. Cette variabilité pose des questions complexes aux médecins. Comment déterminer le bon dosage ? Combien de temps le patient devrait-il suivre le traitement ? Existe-t-il une stratégie optimale propre à un individu ?
Conscients de ces enjeux, nous avons initié un projet ambitieux pour répondre à ces questions. En exploitant les avancées récentes en mathématiques et en informatique, nous développons une application en ligne destinée aux cliniciens. Elle a pour but de fournir des recommandations basées sur des modèles mathématiques complexes qui prennent en compte une multitude de paramètres, des données individuelles du patient aux retours d’expérience globaux sur l’IFNα.
Cette application n’est pas qu’un simple outil de prédiction. Elle est conçue pour être une véritable plateforme d’aide à la décision. Les médecins pourront non seulement obtenir des prévisions sur la réponse probable d’un patient au traitement, mais aussi recevoir des conseils sur l’ajustement des dosages. L’outil vise également à indiquer la durée optimale du traitement, en mettant en balance les bénéfices thérapeutiques et les risques potentiels d’effets secondaires.
ÉTUDES DE BIOLOGIE RELIÉES À DES PROTOCOLES DE RECHERCHE CLINIQUE, DES ÉTUDES TRANSLATIONNELLES OU DES ESSAIS PRÉCOCES

Aline FLOC’H
INFLUENCE DES POLYMORPHISMES DE GROUPES SANGUINS ASSOCIÉS AU SYSTÈME DU COMPLÉMENT DANS L’HÉMOLYSE POST-TRANSFUSIONNEL RETARDÉE DRÉPANOCYTAIRE
La transfusion de globules rouges (GR) est un des traitements principaux de la drépanocytose, première maladie génétique en France. Rarement et de manière presque imprévisible, une destruction des GR transfusées appelée « hémolyse post-transfusionnelle retardée » (HPTR) peut survenir même lorsque les GR transfusées sont tout à fait compatibles. Cette complication rare est sévère car l’hémolyse active le système du « complément » et atteint les organes. Ce système du complément est un acteur incontournable de l’immunité et de la protection contre l’autoimmunité. A la surface des GR, plusieurs groupes sanguins sont portés par des protéines régulatrices du système du complément, dont Cromer (CD55/DAF) et Knops (CR1). Ce projet a pour objectif de rechercher des facteurs de risque de survenue ou de sévérité d’HPTR parmi les groupes sanguins portés par les protéines CD55/DAF et CR1, qui pourraient influencer les effets du système complément dans l’HPTR. Nous comparerons la répartition des groupes sanguins Cromer et Knops entre les patients ayant ou non développé une HPTR. Par une étude de bioinformatique structurale des protéines, nous simulerons le comportement 3D de ces protéines en reproduisant des conditions bioinformatiques proches de celles du GR. Ces analyses nous permettront d’identifier les groupes sanguins des protéines CD55/DAF et CR1 dont nous analyserons ensuite expérimentalement l’impact fonctionnel, afin d’identifier des facteurs de risque ou des facteurs protecteurs de la survenue des HPTR et d’adapter la transfusion et/ou la surveillance des patients drépanocytaires à risque.

Antoine RAUCH
MICROHÉMORRAGIES CÉRÉBRALES DANS LA MALADIE DE WILLEBRAND: PRÉVALENCE ET RETENTISSEMENT COGNITIF WILLMICOG (von WILLbrand disease, cerebral MIcrobleeds and COGnition)
Les micro-saignements cérébraux sont de petites lésions silencieuses qui ne sont vues qu’en imagerie par résonance magnétique (IRM). Leur détection augmente avec l’âge ou dans certaines situations associées à une atteinte des petits vaisseaux cérébraux. Les capacités cérébrales des individus atteints de micro-saignements cérébraux sont diminuées en comparaison aux sujets sains. Une fréquence élevée de micro-saignements cérébraux a été récemment rapporté dans un contexte de maladie hémorragique acquise. Nous avons comme hypothèse que ces micro-saignements cérébraux peuvent également être fréquents dans un contexte de maladie de Willebrand, la plus fréquente des maladies hémorragiques héréditaires. Ce projet de recherche a ainsi pour objectif de déterminer la fréquence et la sévérité des micro-saignements cérébraux en présence d’une maladie de Willebrand et le retentissement de ces lésions sur les capacités cérébrales. Pour cela, nous utiliserons 2 approches complémentaires : 1) une étude clinique qui aura pour objectif d’évaluer la fréquence des micro-saignements cérébraux chez des patients adultes atteints de maladie de Willebrand par la réalisation d’une IRM cérébrale, 2) un modèle de souris génétiquement modifiée permettant de reproduire une forme sévère de maladie de Willebrand où nous induirons des microsaignements cérébraux afin d’étudier leur fréquence et leur retentissement sur la cognition dans un contexte de déficit sévère en facteur Willebrand.

Morgane CHEMINANT
CARACTERISATION CLINICO-BIOLOGIQUE DES LYMPHOMES A CELLULES DU MANTEAU A TRES HAUT RISQUE
Les lymphomes à cellules du manteau (LCM) sont des maladies d’évolution très hétérogène, avec 10 % des patients réfractaires à une chimiothérapie intensive de première ligne. Le pronostic de ces patients est catastrophique, les survies étant inférieures à 2 ans, justifiant l’utilisation de thérapies alternatives innovantes, comme les thérapies ciblées ou les immunothérapies. Actuellement, aucun score ou bio-marqueur ne permet d’identifier ces patients à très haut risque (VHR) et on ne connait pas les déterminants physiopathologiques qui expliquent cette chimiorésistance. Pour cette étude, nous avons sélectionné les patients à très haut risque (10.4 %) d’une large étude française (Le Gouill, Hermine et al, NEJM 2017) chez qui sera réalisée une analyse intégrée des données biologiques et cliniques. Ainsi, les premiers séquençages de l’exome des tumeurs montrent des mutations des voies de TP53 et de réparation de l’ADN ainsi que, de manière surprenante, des mutations impliquées dans la résistance à des thérapies ciblées dès le diagnostic du LCM. Par ailleurs, les variations structurales du génome, qui ont probablement un impact important dans le LCM, sont en cours d’étude par cartographie optique du génome (Bionano Genomics). Le profil d’expression génique par RNA-seq et l’analyse in situ des signatures transcriptomique et protéique tumorales et du micro-environnement permettront d’étudier l’impact de ces mutations, la coexistence éventuelle de sous-populations tumorales différentes, les caractéristiques des cellules tumorales ainsi que ses rapports avec le micro-environnement. L’intégration de ces données nous permettra de discuter l’intérêt d’immunothérapies (cellules T à récepteur chimérique CAR-T et anticorps bispécifiques), de thérapies ciblées ou de combinaisons en première ligne chez ces patients à très haut risque. L’objectif est de proposer une stratégie thérapeutique adaptée dès le diagnostic à ces patients dont l’évolution reste difficile à prédire.

Élise CHAPIRO
LEUCÉMIES LYMPHOÏDES CHRONIQUES AGRESSIVES : RÔLE DU GAIN 2P, DE LA DÉLÉTION 8P ET DES ANOMALIES DU GÈNE MYC DANS LA RÉSISTANCE AUX THÉRAPIES CIBLÉES. ETUDE TEACLL (TWO EIGHT CHROMOSOMAL ABNORMALITIES IN CHRONIC LYMPHOCYTIC LEUKEMIA)
La leucémie lymphoïde chronique (LLC), la leucémie de l’adulte la plus fréquente dans les pays occidentaux, est caractérisée par l’accumulation de lymphocytes B tumoraux dans le sang, la moelle osseuse, la rate et les ganglions. L’évolution est variable : certains patients ont une maladie stable pendant de nombreuses années, alors que d’autres développent des formes agressives nécessitant un traitement. Plusieurs nouvelles molécules sont disponibles pour traiter la LLC, mais malgré les progrès récents, elle reste incurable, avec la survenue de rechutes et de résistance aux traitements. Il existe plusieurs marqueurs génétiques associés aux formes agressives de la maladie qui sont recherchés dans tout bilan pré-thérapeutique. Cependant, d’autres altérations génétiques, moins bien connues car moins fréquentes, sont également retrouvées dans les LLC agressives, parmi lesquelles le gain d’une partie du chromosome 2, la délétion d’une partie du chromosome 8, et des remaniements du chromosome 8 impliquant le gène MYC. Notre projet comprend trois approches complémentaires visant à étudier ces trois anomalies chromosomiques encore peu explorées. Premièrement, grâce à des modèles cellulaires CRISPR/Cas9 développés dans notre laboratoire reproduisant les anomalies chromosomiques concernées, nous chercherons à caractériser la réponse aux différents traitements, et à identifier les voies moléculaires conduisant à l’acquisition de résistance. Deuxièmement, nous travaillerons avec les mêmes objectifs sur des échantillons de cellules de patients. Enfin, avec le suivi clinique de patients porteurs de ces anomalies, nous aurons une analyse précise des patients répondeurs ou non aux traitements « en vraie vie ». Ce projet permettra de mieux comprendre les mécanismes de résistance aux thérapies, avec des retombées attendues sur l’adaptation de la prise en charge des patients.

Lina BENAJIBA
MIEUX COMPRENDRE LES FORMES AGRESSIVES DE LEUCÉMIE POUR IDENTIFIER DE NOUVEAUX TRAITEMENTS
Les néoplasies myéloprolifératives sont une famille de cancers chroniques liés à la multiplication incontrôlée de cellules sanguines au sein de la moelle osseuse. Les traitements actuels ont pour principal objectif d’éviter les complications à court terme et ne permettent pas de guérir la maladie. L’une des complications les plus craintes à long terme est l’évolution de ces pathologies vers une maladie aiguë plus agressive, la leucémie aiguë myéloïde. Les raisons de cette évolution au pronostic catastrophique (<10% de guérison) restent à ce jour largement méconnues. L’objectif principal de nos travaux de recherche est d’identifier de nouvelles approches thérapeutiques ciblées pour traiter ces leucémies agressives, grâce à une compréhension plus fine des voies déréglées à ce stade de la maladie. Pour cela, nous étudierons d’abord en détail les caractéristiques moléculaires de ces leucémies grâce à des échantillons de cellules de patients recueillis à l’échelle nationale dans le cadre d’un réseau de travail spécifique. Ensuite nous chercherons à évaluer l’impact du ciblage des voies déréglées ainsi identifiées sur la progression de la maladie, à l’aide d’un criblage large échelle de molécules chimiques. L’efficacité des traitements les plus prometteurs sera ensuite validée avec pour objectif in fine de développer de nouvelles pistes thérapeutiques à explorer dans le cadre d’essais cliniques nationaux optimisés. Nos travaux devraient ainsi permettre une meilleure compréhension des mécanismes de l’évolution des néoplasies myéloprolifératives vers une maladie plus agressive, et ouvrir la voie vers de nouvelles thérapeutiques qui permettraient d’améliorer le pronostic de cette évolution dévastatrice.
ÉTUDES PORTANT SUR L’ÉPIDÉMIOLOGIE, LA PHARMACO-ÉCONOMIE, L’ÉDUCATION THÉRAPEUTIQUE, LES SCIENCES HUMAINES, LA QUALITÉ DE VIE ET L’ACCOMPAGNEMENT DES PATIENTS

Léa SUREAU
DÉVELOPPEMENT D’UN OUTIL DYNAMIQUE POUR L’IDENTIFICATION DES PATIENTS ATTEINTS DE POLYGLOBULIE DE VAQUEZ ET DE THROMBOCYTÉMIE ESSENTIELLE À HAUT RISQUE D’ÉVOLUTION
Les syndromes myéloprolifératifs sont des leucémies chroniques caractérisées par la production en excès des cellules sanguines (globules rouges, plaquettes ou globules blancs selon les différentes maladies). Ces maladies sont le plus souvent chroniques avec une évolution sur plus de 10 ans mais certains patients peuvent évoluer dans certains cas vers des formes plus graves comme la myélofibrose ou la leucémie aiguë. L’identification des patients à haut risque de complication est actuellement basée sur des paramètres déterminés au moment du diagnostic tels que l’âge, les antécédents de thrombose et le taux des cellules sanguines qui composent des scores dits pronostiques. Certaines études, dont une récente de notre équipe, suggèrent l’intérêt d’une réévaluation dans le suivi du pronostic afin d’affiner l’identification des patients à haut risque d’évolution. Notre projet vise à développer un score pronostique dynamique, c’està- dire applicable tout au long du suivi des patients. Pour cela nous recueillerons les données de suivi de plus de 1 600 patients inclus dans FIMBANK qui est la base clinico-biologique de l’Intergroupe Français des syndromes Myéloprolifératifs (FIM). Le développement de ce score permettra d’identifier au plus tôt les patients à risque d’évolution. L’identification de ces patients est un enjeu majeur dans la prise en charge des syndromes myéloprolifératifs afin d’adapter le suivi et le traitement avec des thérapies ciblées disponibles et en développement dans ces maladies.

Thérèse AURRAN-SCHLEINITZ
BESOIN D’INFORMATION ET DÉCISION MÉDICALE PARTAGÉE DANS LA LLC : POINT DE VUE DES PATIENTS SUR LEUR PRISE EN CHARGE
Une médecine « centrée sur le patient », aujourd’hui reconnue comme améliorant l’état de santé des patient·es, suppose de pouvoir répondre à leurs besoins d’information et leur permettre de prendre des décisions éclairées sur leur maladie en étant le mieux informé possible sur les conséquences de telles ou telles décisions. Si les pathologies chroniques et la cancérologie ont fait l’objet de nombreux travaux, le domaine de l’hématologie est moins étudié et celui de la leucémie lymphoïde chronique (LLC) ne l’est pas. La LLC fait l’objet d’une prise en charge particulière, au cours de laquelle la plupart des patient·es peuvent être seulement surveillé·es sans traitement pendant plusieurs années, établissant ainsi un lien prolongé avec leur hématologue. Deux moments critiques peuvent être identifiés : l’annonce du diagnostic et l’annonce de la nécessité de débuter un traitement. Les progrès thérapeutiques récents offrent de nouveaux traitements d’efficacité équivalente, où les préférences des patient·es peuvent intervenir. L’objectif de ce projet est d’évaluer les besoins d’information de patient·es atteint·es de LLC à l’initiation d’un traitement, et leurs attentes en termes d’implication dans la décision thérapeutique. Ces besoins seront évalués en lien avec le niveau d’anxiété et de dépression des patient·es, leur qualité de vie, et leur niveau socioéconomique. La méthodologie utilisée comprendra à la fois des entretiens et des questionnaires validés dans ce domaine de recherche. Les résultats permettront de construire des supports, à destination des patient.es, d’aide à la compréhension de leur maladie et de leur parcours de soin, d’aide à la décision médicale partagée ainsi qu‘un guide de bonnes pratiques à destination des professionnels de santé.
Etudes portant sur l’épidémiologie, la pharmaco-économie, l’éducation thérapeutique, les sciences humaines, la qualité de vie et l’accompagnement des patients

Jean-Christophe IANOTTO
ENFANTS, ADOLESCENTS ET JEUNES ADULTES PORTEURS DE NÉOPLASIES MYÉLOPROLIFÉRATIVES : ÉTUDE DES CARACTÉRISQUES CLINO-BIOLOGIQUES ET COMPLICATIONS
Les néoplasies myéloprolifératives (NMP) sont des pathologies chroniques de la moelle osseuse associant une augmentation des globules rouges, des plaquettes et/ou des globules blancs. Elles sont liées à l’apparition de mutations moléculaires (bcr-abl, JAK2, CALR ou MPL). Les NMP les plus fréquents sont la polyglobulie de Vaquez, la thrombocytémie essentielle et la myélofibrose primitive. Ces pathologies surviennent le plus souvent après 60 ans et sont associées à des risques vasculaires (thrombose et hémorragie) et évolutifs (myélofibrose et leucémie aigüe). Les patients de plus de 60 ans sont traités par aspirine et cytoréducteur afin de réduire le risque thrombotique. Les patients plus jeunes sont dits moins exposés aux risques de complications et ne sont souvent traités que par aspirine (saignées si polyglobulie). Les patients très jeunes (enfants, adolescents et jeunes adultes) sont rares. Une revue récente de la littérature a montré les limites de nos connaissances de cette population avec l’absence d’informations sur les paramètres clinico-biologiques dans 25 à 80 % des cas. Elle a aussi révélé le manque de suivi qui ne permet pas de connaitre l’incidence réelle des complications classiques, dans cette population. Nous proposons d’utiliser un TEC dédié à cette étude afin de colliger, regrouper et implémenter les données des patients français âgés de 0 à 30 ans (issues des bases de données nationales existantes) au sein d’une seule base. Cette base sera utilisée pour des travaux immédiats d’amélioration de nos connaissances et de nos pratiques concernant ces patients très jeunes atteints de pathologies chroniques et elle servira secondairement de base de départ pour des travaux ambitieux biologiques et thérapeutiques dans cette population peu étudiée.
IMPACT DE 2 STRATÉGIES TRANSFUSIONNELLES SUR LA QUALITÉ DE VIE DES PATIENTS PRÉSENTANT UN SYNDROME MYÉLODYSPLASIQUE DE FAIBLES RISQUES ET MULTITRANSFUSES : ÉTUDE RANDOMISÉE MULTICENTRIQUE COMPARANT UN RÉGIME TRANSFUSIONNEL LIBÉRAL VERSUS RESTRICTIF

Laurent PASCAL
Ce projet vise à comparer l’impact d’un régime transfusionnel « libéral » ( transfusion dès que le patient a moins de 10 g/dL d’hémoglobine) à un régime « Restrictif » classique (transfusion dès que le parient à moins de 8 G/dL d’hémoglobine) sur la qualité de vie à moyen terme des patients présentant un syndrome myéloblastique de faible risque. nécessitant des transfusions régulières en concentrés érythrocytaires. 174 patients seront tirés au sort pour bénéficier de l’un de ces 2 régimes transfusionnels (libéral /restrictif) . La prise en charge des patients est calquée sur la prise en charge habituellement préconisée, seul le seuil transfusionnel étant modifié. Le seuil transfusionnel est déterminé par le groupe de l’étude. Des échelles de qualité de vie et de performance physique des patients sont recueillis. Les incidents transfusionnels sont dénombrés et le coût des transfusions calculés. La période d’observation de l’étude dure 3 ans. L’objectif de ce travail est de démontrer qu’un support transfusionnel plus « libéral » améliore la qualité de vie des patients, sans engendrer de surcoût ni de complication. Ce travail peut avoir un impact sur les politiques de transfusion des patients porteurs d’un syndrome myélodysplasique de faible risque dans tous les services d’hématologie de France.
Biologie-Etudes translationnelles-Essais précoces
ÉTUDES SUR LA PHYSIOPATHOLOGIE DES THROMBOPÉNIES THROMBOTIQUES INDUITES PAR LES VACCINS ANTI-COVID-19 ET ÉVALUATION DE TRAITEMENTS INNOVANTS

Caroline VAYNE
Les thrombopénies thrombotiques induites par un vaccin (VITT) contre la Covid-19 sont une complication exceptionnelle des vaccins à vecteur adénovirus, mais elles sont très graves, avec des thromboses atypiques associées à une activation majeure de l’hémostase. Les VITT semblent dues à des anticorps dirigés contre une protéine plaquettaire, le facteur plaquettaire 4 (FP4). Ces anticorps activent puissamment différentes cellules, dont les plaquettes, et leur spécificité semble différente de celle d’anticorps anti-FP4 responsables des thrombopénies induites par l’héparine (TIH). D’autre part, les thromboses des VITT sont atypiques car souvent multiples et concernent préférentiellement les troncs veineux cérébraux et splanchniques, lesquels sont rarement touchés au cours des TIH. Ceci suggère un processus d’activation cellulaire différent au cours des VITT. Notre laboratoire est le seul à avoir développé plusieurs anticorps monoclonaux anti-FP4 humanisés, notamment 5B9, qui mime les anticorps de TIH, et 1E12, qui possède des propriétés similaires à celles des anticorps de VITT. Grâce à ces outils originaux, notre projet collaboratif, impliquant l’EA 7501 (Tours), l’UMR 1011 (Lille) et l’UMRS 1255 (Strasbourg), doit nous permettre de mieux caractériser les propriétés des anticorps de VITT, de mieux comprendre la physiopathologie des thromboses au cours des VITT, mais aussi d’évaluer l’intérêt d’outils thérapeutiques originaux pour la prise en charge des patients les plus sévèrement atteints.
IDENTIFICATION DES BIOMARQUEURS DE REPONSE À LA 5-AZACYTIDINE DES LYMPHOMES T ANGIOIMMUNOBLASTIQUES

François LEMONNIER
Les lymphomes T angioimmunoblastiques sont des cancers développés à partir de cellules du système immunitaire appelées lymphocytes T. Ils résistent malheureusement fréquemment aux chimiothérapies conventionnelles, il est donc urgent de développer de nouveaux traitements. Ce lymphome est lié à la présence de mutations ayant pour conséquence de modifier la méthylation de l’ADN. Une drogue appelée 5-azacytidine agit en modifiant le niveau de méthylation de l’ADN et pourrait être efficace dans cette maladie. Elle est testée dans le cadre d’un essai clinique chez des patients atteints de lymphome T angioimmunoblastiques. Cependant, certains patients répondent à ce traitement alors que le traitement n’a pas d’effet sur d’autres. Nous souhaitons dans ce projet identifier les marqueurs prédictifs de réponse à la 5-azacytidine dans les lymphomes T angioimmunoblastiques en analysant la moelle osseuse des patients, en recherchant dans les cellules de la moelle les mutations du lymphome. En effet, ces mutations peuvent survenir dans des cellules souches, et la présence de ces mutations dans les cellules souches pourrait influencer la réponse au traitement. Nous allons également analyser les gènes exprimés dans les tumeurs et suivre l’évolution des mutations du lymphome dans le sang des patients et corréler ces paramètres avec la réponse au traitement
RÉPONSE B MÉMOIRE DANS L’ALLO IMMUNISATION ANTI-Jkb CHEZ LES PATIENTS DRÉPANOCYTAIRES

Mathieu MAHEVAS
La drépanocytose est une hémoglobinopathie autosomique récessive affectant des sujets d’origine afro-antillaise, caractérisée par la polymérisation de l’hémoglobine en cas d’hypoxie, d’acidose, de déshydratation érythrocytaire, avec pour conséquences des crises douloureuses et des atteintes chroniques multiviscérales par occlusion microvasculaire. La transfusion reste un support majeur de l’arsenal thérapeutique de cette maladie. Elle n’est cependant pas sans danger, en effet, elle se heurte au polymorphisme des groupes sanguins entre ces patients d’origine afro-antillaise, et les donneurs, essentiellement d’origine européenne. Les anticorps anti-érythrocytaires sont à l’origine d’impasse transfusionnelle, mais surtout d’accidents d’hémolyse post transfusionnelle gravissimes, pouvant mettre en jeu le pronostic vital, et représentant près de 5 % des causes. de décès au cours de la maladie. Un anticorps fréquent est l’anti-Jkb chez ces patients fréquemment Jkb négatif exposés aux globules rouges de donneurs fréquemment Jkb positif. La particularité de cet anticorps est son évanescence rapide après production à l’issue d’une immunisation primaire, mais aussi d’une restimulation. Lorsque l’antécédent d’immunisation n’est pas connu et que la RAI ne le détecte pas en pré transfusionnel, le protocole transfusionnel ne prend pas en compte la phénocompatibilité Jkb, et l’anticorps peut donc être restimulé par des globules rouges Jkb+, et induire une hémolyse transfusionnelle retardée. Dans ce projet, nous nous intéressons aux mécanismes de l’allo immunisation anti-érythrocytaire et plus particulièrement à la réponse mémoire, en utilisant ce modèle d’allo immunisation anti-Jkb. En effet, une meilleure compréhension de cette réponse mémoire pourrait ouvrir des perspectives de détection des patients préalablement immunisés, mais dont le statut n’est pas connu, car ne produisant plus l’anticorps, et des perspectives de prévention d’une réactivation, et ce surtout, lorsque des CGR non compatibles ne sont pas disponibles, ce qui est fréquemment le cas pour ces patients. En effet, la mise en place d’une parfaite compatibilité nécessite des CGR de donneurs de même origine. Dans cette étude, nous souhaitons caractériser dans une cohorte longitudinale de patients allo-immunisés anti-Jkb l’évolution des cellules B mémoires spécifiques générés contre la protéine Jkb au cours du temps et la maturation d’affinité contre cet antigèn
LES MUTATIONS TET2 AU CARREFOUR DU DEVELOPPEMENT ET DE LA FONCTION DES CELLULES NATURAL KILLER DANS LES SYNDROMES MYLODYSPALSIQUES

Nicolas DULPHY
Les syndromes myélodysplasiques (SMD) sont des cancers hématologiques fréquents, touchant principalement les sujets âgés. Ils sont responsables de cytopénies, fatigue et hémorragies, et sont à risque accru d’évolution en leucémie aiguë. Environ 25% des patients SMD sont porteurs de mutations du gène TET2. Les lymphocytes NK jouent un rôle important dans la surveillance et l’élimination des cellules leucémiques. Des travaux préliminaires du laboratoire ont montré que les mutations de TET2, présentes dans le clone tumoral, étaient également retrouvées dans les lymphocytes NK chez ces patients, et étaient corrélées à une altération de leur fonction anti-leucémique. Les mutations TET2 altèrent la production des lymphocytes NK dans la moelle osseuse, conduisant à des cellules NK non fonctionnelles. L’objectif est d’étudier l’impact des mutations TET2 sur la production des cellules NK chez le patient SMD. Pour ce faire, ous allons caractériser des lymphocytes NK, ainsi que tous les intermédiaires cellulaires de différenciation, par des études phénotypiques afin de mettre en évidence les cellules progénitrices différentiellement présentes et affectées chez les patients SMD TET2mut. Par ailleurs, nous allons réaliser des études moléculaires par analyse d’expression de gènes afin de mettre en évidence les voies de régulation des fonctions cellulaires impliquées.
Résultats escomptés : Notre projet ambitionne de comprendre l’impact des mutations du gène TET2 sur la biologie des lymphocytes NK dans un contexte de SMD, participant à leur altération fonctionnelle et favorisant l’échappement tumoral. D’une façon plus large, la compréhension des mécanismes moléculaires en jeu pourrait permettre de les cibler sur le plan thérapeutique.
ÉTUDE DU PROFIL DE CLONALITÉ DANS LES NÉOPLASIES MYÉLOPROLIFÉRATIVES ET RELATION AVEC LES COMPLICATIONS THROMBOTIQUES

Olivier MANSIER
Les néoplasies myéloprolifératives sont des maladies caractérisées par une production excessive de cellules sanguines. Celles-ci résultent de l’apparition de mutations au niveau de l’ADN, la plus fréquente étant la mutation JAK2V617F. La principale complication de ces maladies est l’apparition de caillots dans les vaisseaux (thromboses). A ce jour, peu de marqueurs permettent de prédire le risque de survenue de ces complications. On sait néanmoins que les patients portant la mutation JAK2V617F présentent un risque plus important. Ce risque pourrait être lié à la proportion de cellules sanguines portant la mutation JAK2V617F.
Les études concernant ce dernier point sont néanmoins contradictoires, probablement car les chercheurs ne sont. intéressés qu’aux globules blancs. Différentes études suggèrent que les globules rouges et les plaquettes participent au déclenchement des thromboses au cours des néoplasies myéloprolifératives. Le projet que nous proposons vise à étudier la proportion de cellules portant la mutation JAK2V617F au niveau des différents types de cellules sanguines (globules blancs, globules rouges et plaquettes). Le profil de répartition de la. mutation sera ensuite comparé entre les patients avec thrombose et ceux sans thrombose afin de déterminer si la proportion de globules rouges et/ou de plaquettes mutées est liée au risque thrombotique. Si tel est le cas, nous disposerons d’un nouveau biomarqueur qui permettra d’adapter au mieux la prise en charge des patients et peut-être de mieux évaluer l’efficacité des traitements dont le but est de réduire le risque de thrombose.
Etudes portant sur l’épidémiologie, la pharmaco-économie, l’éducation thérapeutique, les sciences humaines, la qualité de vie et l’accompagnement des patients

Stéphane Giraudier
 Stéphane Giraudier – Étude longitudinale de qualité de vie. « QUALI-VIE » (FIM)
Stéphane Giraudier – Étude longitudinale de qualité de vie. « QUALI-VIE » (FIM)
Un intérêt croissant est porté aux paramètres de qualité de vie et de santé ressentis pour la décision thérapeutique et/ou le suivi de la prise en charge des maladies chroniques, car ils permettent d’apprécier globalement les effets de la maladie et des traitements sur la santé dans son ensemble : physique, mentale et sociale. A ce titre, de nombreux facteurs peuvent altérer la qualité de vie des patients présentant un syndrome myéloprolifératif (SMP) au cours de leur suivi, mais les études de suivi au cours du temps restent peu nombreuses sur le sujet et jusqu’à présent non rapportées en dehors de protocoles thérapeutiques utilisant de nouvelles molécules. Dans ce contexte, la constitution d' »e-cohortes » permettant l’inclusion et le suivi par internet présente de nombreux avantages (grand nombre de patients, réponses instantanées, coûts moindres). Néanmoins, des questions demeurent concernant notamment la représentativité de l’échantillonnage et par conséquent l’exploitation des résultats.
La constitution d’une e-cohorte de patients SMP permet le suivi de la santé perçue et la validation d’échelles de mesure en préservant un niveau de validité satisfaisant. Nous faisons l’hypothèse que la constitution d’une e-cohorte de patients SMP permettra une meilleure prise en charge et un suivi des malades par la prise en compte de la santé ressentie permettant la constitution et la validation de nouvelles échelles de mesure en préservant un niveau de validité satisfaisant.

Aurélie Cabanne-Hamy
Aurélie Cabanne-Hamy – Un observatoire pour les patients traites pour une leucémie aigüe lymphoblastique en rechute ou réfractaire et caractérisée sur le plan oncogénétique. Observatoire ALL-TARGET
Les patients de 60 ans ou plus sont de plus en plus fréquemment allogreffés, notamment pour des maladies hématologiques de pronostic défavorable sans greffe, voire incurables. Ces greffes
se sont développées grâce aux progrès des dernières années induisant une moindre toxicité de la procédure de greffe. Les taux de mortalité non liée à la rechute après allogreffe de cellules souches hématopoïétiques restent cependant élevés entre 30 et 50% et sont influencés par l’âge, les antécédents du patient, la maladie et le type de greffe. Les complications immunitaires, et notamment la réaction du greffon contre l’hôte et les infections survenant après la greffe sont les principales causes de mortalité. L’objet de cette étude est d’évaluer les patients avec des questionnaires « gériatriques » et des tests simples (vitesse de marche) déjà largement utilisés et validés dans la prise en charge des patients avec cancer. Ces évaluations seraient faites avant la greffe et après la greffe en vue d’identifier quels sont les meilleurs marqueurs capables de prédire la mortalité mais aussi la qualité de vie après la greffe. Effectivement, la qualité de vie peut être rapidement altérée du fait d’une dénutrition elle-même secondaire à une infection ou une hospitalisation pour une complication, y compris chez des patients qui seraient en rémission de leur maladie hématologique. Nous avions déjà mis en évidence au sein de notre unité de greffe que les patients les plus âgés sont les plus susceptibles d’être dénutris à J+100 post- greffe.![]() En vue de mettre en place les meilleurs moyens pour lutter contre ces complications et leurs conséquences, nous pensons qu’il est primordial d’identifier les facteurs de risque de mortalité et d’altération de la qualité de vie dans cette population âgés de plus de 60 ans à travers une grande étude prospective multicentrique évaluant de multiples domaines en pré-greffe et post-greffe: capacité physique et cardiaque, autonomie, psychologie, nutrition, insertion sociale, antécédents et maladie chronique.
En vue de mettre en place les meilleurs moyens pour lutter contre ces complications et leurs conséquences, nous pensons qu’il est primordial d’identifier les facteurs de risque de mortalité et d’altération de la qualité de vie dans cette population âgés de plus de 60 ans à travers une grande étude prospective multicentrique évaluant de multiples domaines en pré-greffe et post-greffe: capacité physique et cardiaque, autonomie, psychologie, nutrition, insertion sociale, antécédents et maladie chronique.

Laetitia Largeaud
 Laetitia Largeaud – Impact des facteurs non-cliniques sur les caractéristiques génétiques et la réponse aux traitements dans les leucémies aiguës myéloïdes
Laetitia Largeaud – Impact des facteurs non-cliniques sur les caractéristiques génétiques et la réponse aux traitements dans les leucémies aiguës myéloïdes
Les LAM sont un groupe de maladies hétérogènes avec un mauvais pronostic, influencé à la fois par des facteurs spécifiques au patient et par des facteurs spécifiques à la maladie ou à sa réponse au traitement. En ce qui concerne les facteurs liés à la maladie, de nombreuses études soutiennent que les caractéristiques cytogénétiques et moléculaires sont les facteurs de pronostic les plus puissants. Quant aux caractéristiques des patients, des travaux ont mis en évidence le rôle du contexte social en montrant, en dépit d’un contexte de système de santé subventionné par l’impôt, un accès plus faible à la chimiothérapie et à l’allogreffe chez les patients ayant respectivement un niveau d’éducation plus faible et chez ceux ayant bénéficiant d’un soutien social plus faible. Pourtant le rôle de ces facteurs non-cliniques (FNC) reste mal connu bien que ceux-ci puissent intervenir bien en amont sur le pronostic, en impactant les caractéristiques de la maladie, y compris cytogénétiques et moléculaires par le biais de l’épigénétique. Plusieurs travaux soutiennent en effet que ces mécanismes constituent des voies potentielles reliant les expositions au stress social aux modifications du système immunitaire pouvant favoriser le développement et la progression du cancer. La relation entre la PSE et le paysage moléculaire, la présentation clinique et les résultats des traitements, a rarement été explorée dans le cas du cancer, et jamais dans la LAM. C’est pourquoi, dans ce projet, nous étudions le rôle du contexte social et du milieu de vie (environnement et expositions associées) sur le profil moléculaire et cytogénétique de la LAM à partir d’une large base de données interrégionale. Nos résultats apporteront une meilleure compréhension de l’impact du macro-environnement des patients atteints de LAM sur la biologie de la maladie et la réponse au traitement, mais aussi de documenter l’impact des FNC chez les patients allogreffés ou non.
Biologie-Etudes translationnelles-Essais précoces

Damien Roos-Weil
Damien Roos-Weil – Impact pronostique du paysage moléculaire et micro environnemental de la macroglobulinémie de Waldenström au moment de la mise en route du traitement de première ligne
Les cancers résultent de l’apparition de modifications au sein de l’ADN de cellules aboutissant à leur prolifération incontrôlée. Cette prolifération est également dépendante des cellules dites du microenvironnement qui entourent les cellules cancéreuses. Les mutations de gènes et les anomalies de chromosomes sont parmi les modifications les plus fréquentes à l’origine des cancers. Des mutations de différents gènes peuvent se rencontrer dans un même type de cancer. La présence ou non d’une mutation peut dans de nombreux cancers prédire le pronostic des patients et parfois orienter le choix d’un traitement. La maladie de Waldenström (MW) est un cancer rare d’un sous-type de globules blancs, pour lequel peu de mutations de gènes ayant un rôle pronostique ou dans le choix du traitement ont été décrites.
Nous analyserons les anomalies génétiques (mutations, anomalies chromosomiques) et du microenvironnement d’échantillons d’une population de 110 patients traités pour la première fois pour une MW, avec un traitement équivalent de type immunochimiothérapie. L’objectif de notre travail sera d’identifier les anomalies génétiques associées à un mauvais pronostic après immunochimiothérapie, afin d’envisager ainsi à terme pouvoir proposer aux patients un traitement personnalisé (immunochimiothérapie versus thérapie ciblée) selon leurs caractéristiques génétiques. L’analyse du micro environnement pourra permettre d’identifier des compartiments cellulaires éventuellement responsables de la résistance au traitement et qui pourrait apporter de nouvelles pistes thérapeutiques.

Vincent Camus
 Vincent Camus – Caractérisation biologique et moléculaire complète des lymphomes B du médiastin – Projet PMBL LYSA BIO
Vincent Camus – Caractérisation biologique et moléculaire complète des lymphomes B du médiastin – Projet PMBL LYSA BIO
Le lymphome B du médiastin est un cancer se développant à partir des ganglions lymphatiques et touchant préférentiellement les jeunes adultes, plus particulièrement les femmes. Le diagnostic est difficile du fait des risques à biopsier le médiastin (espace anatomique situé entre les deux poumons et le cœur) et de la complexité d’identifier ce type de lymphome, qui peut être confondu avec un lymphome de Hodgkin ou un lymphome B à grandes cellules. Le lymphome B du médiastin est un cancer agressif pouvant menacer la vie à court terme mais potentiellement curable dans la majorité des cas. Néanmoins, certains patients résistent au traitement ou rechutent très rapidement.
L’équipe de l’unité Inserm U125 rattachée au Centre Henri Becquerel et conjointement avec des médecins et chercheurs du groupe coopérateur LYSA (LYmphoma Study Association) cherche à mieux comprendre ce lymphome en augmentant les connaissances autour de la biologie de ce cancer très particulier pour aider à mieux le diagnostiquer, contribuer au développement de nouvelles cibles thérapeutiques et définir le suivi le plus adapté au risque de rechute.
Pour cela, nous allons réaliser des analyses moléculaires innovantes sur les biopsies archivées de plus de 300 patients qui ont été pris en charges ces dix dernières années pour un lymphome B du médiastin. Nous allons notamment étudier l’ADN et l’ARN de ces tumeurs pour rechercher des anomalies spécifiques, et explorer l’environnement des cellules tumorales, appelé le « micro-environnement ».
Notre étude aura pour impact l’identification de marqueurs d’aide au diagnostic de ce lymphome très rare. Nos résultats permettront également de trouver de nouvelles cibles thérapeutiques qui permettront à terme la recherche de nouveaux traitements, en particulier dans les formes résistantes à la chimiothérapie.

Emmanuelle Clappier
 Emmanuelle Clappier – Oncogenèse des leucémies aiguës lymphoblastiques B du sujet âgé et rôle d’une hématopoïèse clonale
Emmanuelle Clappier – Oncogenèse des leucémies aiguës lymphoblastiques B du sujet âgé et rôle d’une hématopoïèse clonale
Le devenir des adultes atteints de leucémies aigues lymphoblastiques B (LAL-B) reste sombre, en particulier dans la population plus âgée ((≥ 45 ans) qui tolère mal la chimiothérapie intensive. Le développement de nouveaux traitements de type anticorps cibles sur les cellules B a ouvert de nouvelles perspectives pour ces patients. Cependant des phénomènes de rechute sont observés lies
à la capacite des cellules leucémiques à échapper à ces traitements.
Notre projet a pour but de mieux comprendre les particularités des LAL-B du sujet plus âgé, en particulier qui les rendraient plus susceptibles de récidiver après traitement.
Nous faisons l’hypothèse que ces leucémies seraient issues d’une cellule souche, ce qui leur confèrerait une capacite à changer de nature cellulaire pour échapper au traitement cible sur les cellules B.
Pour étayer cette hypothèse nous analyserons des prélèvements de patients afin de mettre en évidence au sein des cellules mutées des marqueurs de cellules souches et la capacité à produire les différents types de cellules sanguines. Nous analyserons également des prélèvements de rechute de patients afin de déterminer le rôle de cette propriété dans la survenue de la rechute.
Au total ces travaux devraient permettre de mieux comprendre ces leucémies, leurs mécanismes de résistance à ces nouveaux traitements et finalement d’optimiser les stratégies de traitement et le suivi des patients.

Raphaël Itzykson
 Raphaël Itzykson – Etude prospective de faisabilité d’un criblage pharmacologique multiparamétrique en conditions pseudo-niche dans les leucémies aiguës myéloïdes
Raphaël Itzykson – Etude prospective de faisabilité d’un criblage pharmacologique multiparamétrique en conditions pseudo-niche dans les leucémies aiguës myéloïdes
Les leucémies aiguës myéloïdes (LAM), les plus fréquentes des leucémies aiguës, restent difficiles à traiter lorsqu’elles rechutent après chimiothérapie. Les médicaments approuvés dans d’autres champs thérapeutiques comme l’oncologie peuvent parfois être repositionnés dans la LAM. Cependant, des combinaisons sont le plus souvent nécessaires pour obtenir une réponse clinique avec de nouveaux agents dans les LAM en rechute.
Un criblage pharmacologique ex vivo à haut débit permet d’identifier les meilleures drogues ou combinaisons, à l’échelle du patient pour permettre une oncologie de précision, et à l’échelle populationnelle pour identifier des combinaisons innovantes et augmenter le taux de réussite de leur transfert en recherche clinique. La faisabilité d’un criblage pharmacologique ex vivo prospectif dans un délai cliniquement pertinent reste cependant mal établie dans les LAM en rechute.
Notre projet s’appuie sur une méthode de criblage pharmacologique à haut débit sur cellules primaires de LAM, combinant de manière innovante des conditions de culture mimant la niche leucémique (stroma, hypoxie et milieu pseudo-plasma) avec une évaluation multiparamétrique de la viabilité, de la différentiation, et du potentiel souche leucémique. Notre projet vise d’abord à établir la faisabilité d’un criblage pharmacologique prospectif dans un délai de 7 jours après prélèvement lors d’une rechute, chez les patients inclus prospectivement dans l’étude PPP (observatoire ALFA). Ensuite, en appliquant pour la première fois à une étude de criblage pharmacologique un design adaptatif bayésien pour tirer le meilleur parti des ressources cellulaires limitées d’un échantillon primaire de patient, notre second objectif est de cribler un portefeuille de 43 médicaments approuvés par l’Agence Européenne du Médicament (EMA) en oncologie pour identifier les 20 molécules les plus actives en monothérapie, puis classer l’activité des 190 combinaisons en résultant, dans une cohorte de 200 patients.
Notre projet établira la faisabilité d’un criblage pharmacologique prospectif selon une technologie innovante. Il permettra d’initier des essais de médecine de précision basés sur ce test. Nous identifierons également de nouvelles combinaisons de médicaments pouvant être évaluées cliniquement.

Damien Luque-Paz
Damien Luque-Paz – Intérêt pronostique des mutations additionnelles dans la polyglobulie de Vaquez et la thrombocytémie essentielle
Les syndromes myéloprolifératifs sont des hémopathies chroniques caractérisées par la production en excès des cellules sanguines (globules rouges, plaquettes ou globules blancs selon les différentes maladies). Ces maladies sont le plus souvent indolentes mais peuvent parfois évoluer vers des formes plus graves comme la myélofibrose ou la leucémie aigüe. L’origine de ces pathologies est liée a l’apparition de mutations de l’ADN des cellules de la moelle osseuse ou sont produites toutes les cellules du sang. Ces mutations, découvertes depuis 2005, touchent les gènes JAK2, CALR ouMPL et sont à l’origine de la production en excès des cellules sanguines. Les progrès technologiques du séquençage de l’ADN rendent aujourd’hui possible l’analyse rapide, sensible et simultanée d’un grand nombre de gènes. De cette façon, d’autres mutations de l’ADN, dites additionnelles, peuvent être détectées dans les syndromes myéloprolifératifs. Certaines de ces mutations semblent avoir un rôle dans l’évolution de la maladie et leur détection permettrait d’identifier au plus tôt les patients à haut risque d’évolution défavorable. Toutefois, les études sur un grand nombre de patients sont encore peu nombreuses et l’identification précise des mutations de mauvais pronostic reste à préciser.
Notre projet vise à étudier 500 patients par séquençage haut-débit de 77 gènes afin d’identifier des signatures génétiques associées a un risque d’évolution. Les patients seront issus de FIMBANK qui est la base clinico-biologique de l’Intergroupe Francais des syndromes Myéloprolifératifs (FIM). Les données générées permettront également d’étudier précisément le rôle de certains traitements sur le pronostic confère par les mutations additionnelles. Les résultats de ce projet pourront conduire à une définition consensuelle des mutations à haut risque d’évolution dans les syndromes myéloprolifératifs. L’identification de ces patients est un enjeu majeur dans la prise en charge des syndromes myéloprolifératifs afin d’adapter le suivi et le traitement avec des thérapies ciblées disponibles et en développement dans ces maladies.
Etudes portant sur l’épidémiologie, la pharmaco-économie, l’éducation thérapeutique, les sciences humaines, la qualité de vie et l’accompagnement des patients

Lilas Gillis
 Lilas Gillis – Etude prospective observationnelle, multicentrique de l’impact sur la relation parent/enfant(s) d’un robot de téléprésence mobile au domicile de patients ayant des enfants de moins de 15 ans et hospitalisés en isolement protecteur de longue durée (« VIK-e 2 »)
Lilas Gillis – Etude prospective observationnelle, multicentrique de l’impact sur la relation parent/enfant(s) d’un robot de téléprésence mobile au domicile de patients ayant des enfants de moins de 15 ans et hospitalisés en isolement protecteur de longue durée (« VIK-e 2 »)
Les traitements intensifs de certains cancers et hémopathies nécessitent pour le patient de longues périodes d’isolement en chambre stérile allant jusqu’à plusieurs mois. Dans certains services, les visites sont interdites aux enfants en bas âge pour limiter le risque de transmission microbienne. En septembre 2016, afin de maintenir le lien social entre les jeunes hospitalisés et leurs proches, les professionnels de santé de l’Institut d’Hématologie et d’Oncologie Pédiatrique (Lyon) ont expérimenté une nouvelle technologie : le robot de téléprésence (projet VIK-e). Le principe repose sur un ordinateur installé dans la chambre du patient qui commande à distance un robot mobile localisé à son domicile, permettant ainsi d’échanger avec les personnes présentes Cette étude a mis en évidence que le robot permet le maintien d’un lien avec la fratrie par la communication mais également par des interactions plus ludiques et par la participation à des moments clés de la vie familiale. Les communications par robot sont décrites comme moins conventionnelles et plus spontanées qu’avec les autres outils de communication (type Skype). Le robot est perçu comme une source de mieux être et semble aider les patients à mieux vivre l’hospitalisation et l’isolement. Nous avons, dans le cadre de ce projet, « expérimenté » ce robot chez une jeune maman de deux petits garçons et avons de suite constaté l’utilisation enthousiaste qu’elle faisait de ce robot, allant réveiller tous les matins ses enfants et leur racontant une histoire avant d’aller se coucher. Très vite, nous nous sommes demandé si l’utilisation du robot de téléprésence ne pourrait pas avoir des bénéfices tout aussi importants, chez les patients parents de jeune(s) enfant(s). Nous avons donc décidé de monter le projet « VIK-e 2 » en proposant aux patients parents de jeune(s) enfant(s) de bénéficier du robot de téléprésence lors de leur hospitalisation en chambre stérile et en évaluant son intérêt dans le maintien de la relation parent/enfant(s).

Marie Robin
Marie Robin – Evaluation multi-domaine et qualité de vie des patients de 60 ans ou plus recevant une allogreffe de cellules souches hématopoïétiques (CSH)
Les patients de 60 ans ou plus sont de plus en plus fréquemment allogreffés, notamment pour des maladies hématologiques de pronostic défavorable sans greffe, voire incurables. Ces greffes
se sont développées grâce aux progrès des dernières années induisant une moindre toxicité de la procédure de greffe. Les taux de mortalité non liée à la rechute après allogreffe de cellules souches hématopoïétiques restent cependant élevés entre 30 et 50% et sont influencés par l’âge, les antécédents du patient, la maladie et le type de greffe. Les complications immunitaires, et notamment la réaction du greffon contre l’hôte et les infections survenant après la greffe sont les principales causes de mortalité. L’objet de cette étude est d’évaluer les patients avec des questionnaires « gériatriques » et des tests simples (vitesse de marche) déjà largement utilisés et validés dans la prise en charge des patients avec cancer. Ces évaluations seraient faites avant la greffe et après la greffe en vue d’identifier quels sont les meilleurs marqueurs capables de prédire la mortalité mais aussi la qualité de vie après la greffe. Effectivement, la qualité de vie peut être rapidement altérée du fait d’une dénutrition elle-même secondaire à une infection ou une hospitalisation pour une complication, y compris chez des patients qui seraient en rémission de leur maladie hématologique. Nous avions déjà mis en évidence au sein de notre unité de greffe que les patients les plus âgés sont les plus susceptibles d’être dénutris à J+100 post- greffe. En vue de mettre en place les meilleurs moyens pour lutter contre ces complications et leurs conséquences, nous pensons qu’il est primordial d’identifier les facteurs de risque de mortalité et d’altération de la qualité de vie dans cette population âgés de plus de 60 ans à travers une grande étude prospective multicentrique évaluant de multiples domaines en pré-greffe et post-greffe: capacité physique et cardiaque, autonomie, psychologie, nutrition, insertion sociale, antécédents et maladie chronique.
En vue de mettre en place les meilleurs moyens pour lutter contre ces complications et leurs conséquences, nous pensons qu’il est primordial d’identifier les facteurs de risque de mortalité et d’altération de la qualité de vie dans cette population âgés de plus de 60 ans à travers une grande étude prospective multicentrique évaluant de multiples domaines en pré-greffe et post-greffe: capacité physique et cardiaque, autonomie, psychologie, nutrition, insertion sociale, antécédents et maladie chronique.
Biologie-Etudes translationnelles-Essais précoces

Pierre-Yves Dumas
Pierre-Yves Dumas – Etude d’un épissage alternatif du FLT3 comme marqueur de maladie résiduelle en RT-qPCR dans les LAM
Ces dernières années, les avancées dans l’identification des anomalies moléculaires et cytogénétiques dans les leucémies aiguës myéloblastiques (LAM) ont été considérables et certains marqueurs peuvent être suivis en maladie résiduelle pour déterminer plus précisément le niveau de la réponse aux traitements et donc adapter l’intensité de ces derniers. Des travaux conjoints entre chercheurs et médecins ont permis de décrire une nouvelle forme d’une protéine reconnue comme très importantes dans les leucémies aiguës myéloblastiques. Cette nouvelle anomalie serait présente chez 40% des patients et nous pensons que nous pouvons affiner les stratégies thérapeutiques des patients en suivant son expression au cours du traitement. L’étude proposée ici vise à prouver cette théorie dans un grand essai clinique français.

Stéphanie Nguyen
Stéphanie Nguyen – Transplantation de microbiote fécal dans la prévention de la réaction du greffon contre l’hôte après allogreffe de cellules souches hématopoïétiques pour une hémopathie maligne
La greffe de moelle osseuse reste le seul traitement curateur pour certaines maladies hématologiques mais s’accompagne de complications infectieuses ou immunologiques. Notamment, les cellules du donneur peuvent attaquer les tissus du receveur et déclencher une réaction inflammatoire appelée GVHD= Graft versus Host Disease. Il existe un lien étroit entre les perturbations du microbiote du patient et la GVH ou les infections. Le microbiote (ou flore intestinale) est l’ensemble des microbes présents dans le tube digestif. Il est considérablement perturbé par les chimio/radiothérapies ainsi que l’utilisation des antibiotiques à large spectre utilisés lors de la greffe de moelle. Après des années passées à tenter de « stériliser » au maximum le tube digestif des patients greffés de moelle ; nous savons à présent qu’il est nécessaire de préserver leur microbiote intestinal. Un protocole national français « TMF-Allo » a obtenu un financement visant à évaluer l’effet de la transplantation de microbiote fécal (TMF) chez les patients greffés de moelle. Les patients seront randomisés (tirage au sort) pour recevoir ou pas une TMF provenant d’une banque de donneurs sains et administrée par lavement à 1 mois de la greffe de moelle. L’objectif principal est de voir si la TMF permet d’améliorer la flore intestinale des patients greffés et d’étudier ses effets sur leur devenir (survie, infections, GVH, rechute de l’hémopathie). Ce protocole associe la majorité des centres de greffes adultes français et est le premier protocole au monde posant la question de l’impact de la TMF sur le devenir des patients allogreffés de moelle. Il est essentiel d’associer une étude biologique du microbiote des patients qui auront reçu ou pas une TMF. Nous étudierons la composition bactérienne et virale des selles ainsi que les substances produites par les bactéries (métabolisme) ainsi que les rapports entre le microbiote et la production d’anticorps antibactériens. Seule la partie sur le métabolisme bactérien est présentée à l’AAP Force Hémato.

Bruno Cassinat
Bruno Cassinat – Analyse en cellules uniques de l’évolution clonale de patients avec myélofibrose inclus dans l’essai RUXOPEG
Les néoplasies myéloprolifératives (NMPs) sont des maladies hématologiques malignes chroniques pouvant évoluer dans 20 à 30% des cas vers des formes beaucoup plus graves comme la myélofibrose ou la leucémie aiguë. Il n’existe pas de traitement curatif des NMPs en dehors de la greffe de moelle osseuse qui ne peut être proposée qu’à peu de patients. On sait que l’origine des NMPs vient de l’acquisition de mutations dans certains gènes comme le gène JAK2. Notre équipe a montré que le traitement par interféron alpha (IFNa) peut induire des rémissions prolongées et que c’est le seul traitement capable de réduire significativement le nombre de cellules mutées pour JAK2 (réponse moléculaire). Cependant les patients sont hétérogènes en termes de réponses cliniques et moléculaires. L’origine de cette hétérogénéité
est mal comprise mais de nombreux arguments (venant de notre équipe et d’autres) suggèrent que cela pourrait être dû à l’acquisition de mutations supplémentaires dans d’autres gènes (on parle de mutations additionnelles). Les méthodes de séquençages modernes (NGS) ont permis de montrer que les patients peuvent héberger depuis 1 jusqu’à 5 ou 10 mutations différentes. Mais on sait que ces mutations peuvent être présentes soit dans les mêmes cellules que la mutation de JAK2, soit dans des cellules différentes. On parle de « clones » différents lorsque les mutations sont différentes entre 2 groupes de cellules. Notre projet vise à utiliser une méthodologie très innovante pour étudier à l’échelle unicellulaire la répartition des mutations afin de définir la répartition des clones chez les patients. Nous appliquerons cette technologie à des patients inclus dans un protocole thérapeutique appelé RUXOPEG qui associe, chez des patients atteints de myélofibrose, un traitement par IFNa et un inhibiteur de JAK2 (le ruxolitinib). Ceci nous permettra d’étudier quels types de cellules (caractérisées par les mutations présentes dans celles-ci) sont les plus sensibles ou résistantes au traitement. Ceci permettra de mieux comprendre les mécanismes d’action et de prédire les patients bons ou mauvais répondeurs à ces traitements.
On parle de « clones » différents lorsque les mutations sont différentes entre 2 groupes de cellules. Notre projet vise à utiliser une méthodologie très innovante pour étudier à l’échelle unicellulaire la répartition des mutations afin de définir la répartition des clones chez les patients. Nous appliquerons cette technologie à des patients inclus dans un protocole thérapeutique appelé RUXOPEG qui associe, chez des patients atteints de myélofibrose, un traitement par IFNa et un inhibiteur de JAK2 (le ruxolitinib). Ceci nous permettra d’étudier quels types de cellules (caractérisées par les mutations présentes dans celles-ci) sont les plus sensibles ou résistantes au traitement. Ceci permettra de mieux comprendre les mécanismes d’action et de prédire les patients bons ou mauvais répondeurs à ces traitements.

Olivier Kosmider
Olivier Kosmider – Un biomarqueur de l’érythropoïèse clonale des syndromes myélodysplasiques appliqué aux protocoles du GFM
Les syndromes myélodysplasiques (SMD) sont des cancers du sang qui affectent les sujets âgés et dont l’apparition est reliée a des anomalies génétiques qui touchent les cellules souches de la moelle osseuse. Il existe cependant de nombreux sous types de SMD dont la prise en charge n’est pas toujours identique et pour lesquels il n’est pas facile d’évaluer la réponse à un traitement en dehors de l’utilisation de paramètres biologiques traditionnels mais peu spécifiques. Parmi ces catégories, les SMD avec sidéroblastes en couronne sont réunis dans une catégorie qui regroupe des patients présentant une tendance à une accumulation toxique de fer dans l’organisme et, dans plus de 90% des cas, une anomalie génétique modifiant la fonction d’un gène clé impliqué dans la production des protéines de la cellule, le gène SF3B1. Ainsi modifié, le gène SF3B1 mutant entraine la production de protéines anormales, perturbant le destin de cellules de la moelle osseuse. Notre équipe a récemment montré que les anomalies de SF3B1 entraînaient la création d’une forme anormale d’une protéine récemment identifiée et qui présente un rôle clé dans le métabolisme du fer appelée Erythroferrone ou ERFE. Se basant sur des données préliminaires, notre projet actuel tend à évaluer la pertinence de cette forme anormale d’ERFE en tant que marqueur permettant de suivre l’efficacité des traitements proposés aux patients présentant un SMD avec sidéroblastes en couronnes et mutation de SF3B1.
Notre équipe a récemment montré que les anomalies de SF3B1 entraînaient la création d’une forme anormale d’une protéine récemment identifiée et qui présente un rôle clé dans le métabolisme du fer appelée Erythroferrone ou ERFE. Se basant sur des données préliminaires, notre projet actuel tend à évaluer la pertinence de cette forme anormale d’ERFE en tant que marqueur permettant de suivre l’efficacité des traitements proposés aux patients présentant un SMD avec sidéroblastes en couronnes et mutation de SF3B1.
Etudes portant sur l’épidémiologie, la pharmaco-économie, l’éducation thérapeutique, les sciences humaines, la qualité de vie et l’accompagnement des patients

Loïc Ysebaert

Fabien Despas
![]()
Loïc Ysebaert – Fabien Despas – Qualité de vie des Lymphomes et parcours de soins oncologique à 1 an post-chimiothérapie : QUALYPSO1
La période qui suit la fin de la chimiothérapie pour un lymphome malin (LM), alors que la maladie est en rémission complète et donc simplement surveillée selon des modalités de parcours de soins d’oncologie (PSO) très divers parce que non codifiés précisément, est très peu étudiée. C’est pourtant pour le patient et ses aidants une période tourmentée dans la moitié des cas : problèmes psychologiques (fatigue, peur de la rechute, symptômes anxio-dépressifs variés), sociaux (perte d’argent, perte du travail, sentiment de déclassement), mais aussi troubles physiques séquellaires des traitements reçus (troubles infectieux, digestifs, cardiaques, douleurs, rhumatologiques). Notre équipe à l’IUC de Toulouse travaille depuis 2006 sur des projets de recherche d’accompagnement des malades et de leur famille (Assistance des Malades Ambulatoires, AMA). Elle a la meilleure expérience en France notamment depuis 2012 dans le suivi par un modèle original de « parcours de soins partagés », centré sur le généraliste et une infirmière dédiée à l’après-cancer (AMA-AC, >340 patients inclus à Toulouse), en lien avec l’hématologue référent. Ensemble, avec des échelles de mesures spécifiques validées, ils vont explorer le « retour à la vie sans chimiothérapie », qui n’est jamais un « retour à la vie d’avant », pour mieux appréhender les troubles psychiques, physiques, sociaux et les combattre. L’étude que nous proposons sous l’égide du LYSA consiste à utiliser nos échelles de mesure sur une population de patients traités à travers toute la France, à la fin de la chimio et à un an post-traitement, pour apprécier l’évolution bonne ou mauvaise des troubles constatés, et éventuellement d’identifier les PSO qui diminuent le risque d’altération prolongée de la qualité de vie des patients en rémission de leur LM.

Pascale Cony-Makhoul

Pascale Cony-Makhoul – Rôle du lieu de la prise en charge de l’expertise clinique sur la qualité de la réponse au traitement par l’inhibiteur de tyrosine kinase dans la leucémie myéloïde chronique – Etude exhaustive à partir des registres de population entre 2006 et 2015
La Leucémie Myéloïde Chronique est une maladie rare (environ 800 nouveaux cas diagnostiqués chaque année en France) dont le pronostic a été transformé depuis le début des années 2000 grâce à la mise à disposition du premier traitement oral ciblé (c’est-à-dire qui bloque un élément spécifique à l’origine de la maladie). Cette maladie autrefois constamment mortelle après quelques années d’évolution, est devenue chronique ; il a été montré que ces patients ont une espérance de vie similaire à celle de la population non malade, à la condition de poursuivre le traitement de façon assidue et très prolongée, avec une surveillance régulière assurée par un spécialiste, apte à contrôler la réponse au traitement et à le modifier si toutefois il n’était plus ou pas assez efficace. Le suivi du marqueur de la maladie (transcrit bcr-abl) se fait sur une prise de sang, pour l’instant réalisée dans un nombre limité de laboratoires principalement hospitaliers. La conséquence de ces progrès prodigieux est la nette augmentation du nombre de patients vivants avec cette maladie (qui a presque triplé depuis 2000) ce qui impose de réfléchir à de nouvelles organisations de la prise en charge des patients reposant sur des réseaux régionaux de soins en veillant à ce que la qualité des résultats reste excellente. En France il existe un réseau de registres qui assurent la surveillance des cancers dans des zones géographiquement définies. Ces registres sont des sources de données (démographiques, circonstances de diagnostic, caractéristiques des maladies, parcours de soins, nature et résultats des traitement …), exhaustives, permettant de générer des informations applicables à l’échelle nationale. Cette étude s’appuie sur les 3 registres français des hémopathies malignes et a pour objectif principal de vérifier si le lieu de la prise en charge des patients influence la qualité de la réponse au traitement. Nous espérons pouvoir proposer une graduation et une optimisation des parcours de soins avec le souci de préservation des résultats et d’équité d’accès aux soins.
Biologie-Etudes translationnelles-Essais précoces

Romain Guieze
![]()
Romain Guieze – Histoire génétique de la transformation de la leucémie lymphoïde chronique en syndrome de Richter
La leucémie lymphoïde chronique (LLC) est la forme la plus fréquente de leucémie en Occident. Cette maladie indolente peut parfois évoluer vers un lymphome agressif, appelé syndrome de Richter (SR) dont le pronostic est très défavorable. Alors que la LLC a récemment fait l’objet d’importants progrès (caractérisation des gènes altérés, développement de traitements ciblés), le SR reste peu exploré et aucun des traitements les plus récents ne s’avèrent efficaces. Notre projet a pour objectif de mieux comprendre les mécanismes à l’origine de la transformation de LLC en SR. Par séquençage de l’ADN de l’ensemble du génome codant, nous caractériserons en parallèle les altérations génétiques acquises dans les cellules tumorales du SR et de LLC chez un même patient. Nous analyserons les échantillons tumoraux de 35 patients inclus dans un essai clinique évaluant une nouvelle approche d’immunothérapie (le blinatumomab). Ce travail identifiera les évènements génétiques capable d’induire un SR à partir d’une LLC. Ces informations permettront ultérieurement de modéliser le SR pour mieux l’étudier et révéler de nouvelles stratégies thérapeutiques. Il permettra aussi d’identifier des facteurs pronostiques de réponse à l’immunothérapie. Une meilleure prise en charge du SR permettrait de lever l’un des derniers obstacles au contrôle à long terme de la LLC.

Franck-Emmanuel Nicolini
Franck-Emmanuel Nicolini – PETALs ddPCR – PEgylated interferon-a2a and TAsigna® for first-Line therapy of chronic phase CML patientS digital droplet PCR
Le traitement de la Leucémie Myéloïde Chronique (LMC) a été révolutionné il y a maintenant 20 ans par l’introduction des traitements ciblés qui éliminent de manière sélective les cellules leucémiques. Cependant malgré l’efficacité remarquable de ces médicaments administrés par voir orale, la plupart des malades gardent un contingent stable de cellules leucémiques dans leur corps, ce qui nécessite la poursuite indéfinie de ces agents médicamenteux, ce qui peut être source d’effets indésirables à long voire à très long-terme. Cependant un petit nombre de patient peut accéder à l’arrêt définitif du médicament sans que sa leucémie reprenne un cours évolutif agressif. Pour répondre à cette problématique le groupe français de la LMC (Groupe Fi-LMC) a développé un essai thérapeutique national dans la plupart des services d’hématologie des hôpitaux universitaires de France, l’essai PETALs qui compare un médicament puissant le Tasigna® pour la moitié des patients à ce même médicament associé à un ancien traitement de la maladie, l’interféron sous une nouvelle forme bien mieux tolérée et administrée en piqures.
Les résultats préliminaires de cet essai thérapeutique ou plus de 200 patients ont participé, indépendant de l’industrie, mais soutenu par elle, lancé en 2014 indiquent que le groupe de patients ayant reçu l’association Tasigna® + Interféron répond mieux, plus profondément. Il est possible que le nombre de patients atteignant les critères définitifs d’arrêt des 2 médicaments soit plus élevés également mais ceci est en cours de suivi. Une nouvelle technique de biologie moléculaire permettant la surveillance de la maladie, la PCR digitale (ou ddPCR) est en cours d’évaluation et permet de mesurer très finement la persistance ou non de cellules leucémiques dans le sang des patients atteints de cette leucémie, et nous pensons qu’elle permettrait de mieux discerner les patients qui répondent de manière excellente -et donc susceptibles d’arrêter définitivement et en toute sécurité leurs traitements- par rapport à ceux qui ne doivent pas arrêter leur traitement en raison d’une maladie toujours bien présente. L’objectif de ce projet et de réanalyser tous les malades atteints de cette leucémie candidats à l’arrêt de traitement sur les critères actuels reposant sur la technique de biologie moléculaire de routine (dite RQ-PCR), par cette nouvelle méthode très sensible qu’est la ddPCR et d’évaluer dans ce contexte son utilité et sa fiabilité.

Ludovic Lhermitte
![]()
Ludovic Lhermitte – Co-inhibition de la voie IL7R-Jak-STAT et de Bcl2 dans les leucémies aigues lymphoblatique T de l’adolescent et du jeune adulte
Les leucémies aigües lymphoblastiques T (LAL-T) représentent une expansion de lymphocytes T immatures bloqués dans leur différenciation. Malgré les progrès thérapeutiques, cette pathologie touchant préférentiellement les adolescents et jeunes adultes (AJA) est associée à un mauvais pronostic. C’est le cas en particulier des LAL immature/ETP (Early T-cell Progenitor) qui se caractérisent par un profil clinique particulier (cortico-résistance, chimiorésistance, fréquence des rechutes). Les LAL immatures/ETP sont fréquemment associées à des mutations de la voie de signalisation IL7R-Jak-STAT et à une dépendance à une protéine empêchant la mort cellulaire, Bcl2. La littérature suggère l’intérêt d’inhiber Jak dans les LAL portant une mutation IL7R-Jak-STAT. De manière intéressante, nos travaux (données non publiées) montrent que même en l’absence de mutation, la signalisation IL7R-Jak-STAT est anormale et peut être efficacement inhibée par le médicament ruxolitinib pour induire la mort de la tumeur.
Ce traitement pourrait par conséquent être étendu aux cas de leucémies exprimant l’IL7R indépendamment de la présence ou non de mutation sur la voie IL7R-JakSTAT. Cependant, il existe des cas de résistance primaire ou de sensibilité intermédiaire qu’il convient de contrecarrer. Dans ces cas, nous avons remarqué un profil anormal d’expression de Bcl2 et molécules apparentées (famille BH3) ne répondant pas à l’inhibition de Jak. L’objectif de ce travail s’articule autour de plusieurs axes : i) Déterminer la signature de la voie IL7R-Jak-STAT dans le contexte pathologique des LAL-T, ii)Explorer le profil d’expression des protéines de la famille BH3 en fonction de l’expression de l’IL7R et des mutations de la voie IL7R-Jak-STAT, iii) Analyser in vitro la sensibilité des lymphoblastes en présence d’inhibiteur de Jak, de Bcl2 ou d’une combinaison des 2 pour rechercher une levée de résistance ou une synergie, iv) Analyser in vivo chez la souris leucémique la réponse au traitement conjoint anti-Jak et anti-Bcl2. Les LAL-T ETP affectent les adolescents/adultes jeunes et sont associées à un pronostic défavorable. De nouveaux traitements sont indispensables pour en améliorer le pronostic. Le dogme actuel tend à privilégier les thérapies ciblées, c’est-à-dire à agir sur une protéine dont la population leucémique dépend. Nos travaux précédents montrent l’intérêt de combiner ces stratégies pour ‘attaquer la population leucémique’ sur plusieurs fronts. Ce modèle de thérapie multi cible convergente représente une stratégie originale pour améliorer le pronostic des LAL ETP des AJA.

Emmanuel Bachy
![]()
Emmanuel Bachy – LEIA – Utilisation d’algorithmes d’intelligence artificielle et d’analyses épigénétiques pan-génomiques pour l’amélioration du diagnostic des lymphomes T matures
Les lymphomes non hodgkiniens (LNH) représentent la 6ème cause de cancer en France. Bien qu’ils ne constituent que 15% environ des LNH, les lymphomes T périphériques (LTP) sont l’un des sous-types associés à la mortalité la plus élevée. Non seulement ils sont particulièrement chimio réfractaires, mais ils sont également largement sous-étudiés tant sur le plan de la recherche fondamentale que de la recherche clinique. Ils restent à l’heure actuelle moins bien compris que les autres lymphomes en termes de physiopathologie, d’identification de la cellule tumorale d’origine ou de classification précise. Dans le cadre de ce projet, nous proposons d’utiliser une technique récemment développée et dénommée ATAC-seq (assay for transposase-accessible chromatin using sequencing). Elle permet de répertorier les régions accessibles de la chromatine à l’échelle pan-génomique par séquençage haut débit, sorte de code barre génétique à plusieurs centaines de milliers de barres. L’objectif est d’évaluer le potentiel de cette technique pour la classification précise et rapide (<24-48H) des LTP à un coût largement inférieur aux autres techniques. Avec une collection de plus de 700 échantillons rétrospectifs déjà disponibles, nous pensons proposer en moins de 24 mois une signature spécifique de chaque sous-type de LTP à l’aide d’algorithmes d’intelligence artificielle. Pour conclure, à la lumière des données de la littérature et des données préliminaires obtenues dans notre équipe, nous espérons pouvoir démontrer comment l’ATAC-seq présente une opportunité inégalée de modifier la pratique courante en hématologie.

Christian Recher
Christian Recher – Etude de phase 2 évaluant l’addition de la dexaméthasone à la chimiothérapie d’induction et de consolidation chez les sujets âgés traités pour une leucémie aiguë myéloïde nouvellement diagnostiquée (LAM-SA 2017/DEXAML-02)
La chimiothérapie des patients atteints de leucémie aiguë myéloïde (LAM) n’a pas réellement changé depuis près de 40 ans, combinant deux drogues, la cytarabine et une anthracycline. Les progrès thérapeutiques effectuées au cours du temps restent limités chez les sujets de plus de 60 ans et sont essentiellement liés à l’amélioration des soins de supports, à une meilleure compréhension du pronostic grâce à de nouveaux outils moléculaires et l’extension des indications de greffe de cellules souches hématopoïétiques. Des résultats récents issus d’un centre du groupe FILO ont suggéré que la dexaméthasone, un médicament utilisé de longue date dans d’autres hémopathies, pourrait avoir un rôle important pour la prévention des rechutes en combinaison avec la chimiothérapie. En effet, cette étude a montré que l’utilisation de la dexaméthasone était significativement associée à une réduction de l’incidence de la rechute et à une amélioration de la survie globale chez des patients ayant une LAM avec une hyperleucocytose importante. Ces résultats ont été complétés par données précliniques démontrant que la dexaméthasone sensibilise les cellules souches leucémiques à la chimiothérapie et ainsi limite le risque de croissance leucémique et de rechute. L’essai clinique prospectif soutenu par Force Hémato a pour but d’évaluer si la dexaméthasone ajoutée à la chimiothérapie d’induction et de consolidation va améliorer réellement le devenir des patients en comparaison à l’expérience historique du groupe FILO. Si l’étude est concluante, elle permettra un changement très rapide des pratiques de routine de façon efficace, sûre, facilement diffusable et très peu coûteuse.

Chloé James
Chloé James – Evaluation de nouveaux biomarqueurs de thrombose dans les néoplasies myéloprolifératives
Les néoplasies myéloprolifératifs (NMP) sont des maladies acquises au cours de la vie caractérisées par une production anormalement élevée de cellules sanguines. La complication la plus fréquente chez ces patients est la formation de caillots dans les vaisseaux (thrombose) qui est la principale cause de décès. L’augmentation du nombre de globules blancs est un facteur de risque de thrombose dans ces maladies, sans que l’on ne sache comment. Des études ont montré que les polynucléaires neutrophiles (PNN), qui sont les globules blancs les plus abondants du sang, pouvaient émettre des filets d’ADN, appelés NETs (Neutrophil Extracellular Trap) qui pourraient avoir un rôle dans la thrombose. L’objectif de ce travail est de savoir si des biomarqueurs de NET corrèlent avec la survenue de la thrombose dans les NMP. Cette étude nous permettra d’avancer dans la compréhension de la thromboe chez les patients mais surtout nous permettra de savoir si les biomarqueurs de NETs pourraient être utiles dans la détermination du risque de thrombose, principal risque chez ces patients.

Florence Cymbalista
Florence Cymbalista – Tolérance et résistance à l’ibrutinib chez les patients atteints de leucémie lymphoïde chronique
Les traitements de la leucémie lymphoïde chronique ont beaucoup évolué ces dernières années. Des patients qui n’avaient pas une bonne réponse à la chimiothérapie ont pu bénéficier de nouveaux traitements dit « ciblés » car dirigés vers la machinerie de la cellule malade et donc ne détruisant pas les cellules saines. La France a eu l’autorisation de traiter des patients avec ce nouveau médicament (ibrutinib) depuis maintenant 3 ans. Aux Etats-Unis, où le recul est un peu plus long, la survenue de cas de résistance à ces traitements à pu être observée, dans un délai d’environ 3 ans. Ces résistances sont liées notamment à des mutations dans les cellules malades qui empêchent le médicament d’agir. Ces mutations semblent précéder la survenue de rechute de plusieurs mois. Notre étude consiste donc à rechercher ces mutations avant qu’elles n’aient un impact visible chez les patients traités par ce médicament depuis 3 ans dans 24 centres du groupe FILO participant à l’étude. Ainsi, nous pourrons analyser chez quels patients ces mutations surviennent, et envisager précocement une modification de traitement.
Nicolas Dulphy – LIM-SDM : études des lymphocytes innées médullaires dans les syndromes myélodysplasiques
La moelle osseuse est le site privilégié de production des cellules du système immunitaire, et notamment des populations cellulaires appelées « Natural Killer (NK) », capables de tuer des cellules leucémiques, et « Innate Lymphoid Cells (ILCs) » dont le rôle est de contrôler la qualité de la réponse immunitaire dans les tissus. Cependant, la moelle osseuse est aussi le site de développement de cancers hématologiques tels que les syndromes myélodysplasiques (SMD). Cette maladie peut se transformer, dans sa forme la plus grave, en leucémie aiguë myéloblastique (LAM), et peut aussi induire des maladies auto-immunes. Notre hypothèse est que la colonisation de la moelle par les cellules cancéreuses SMD perturbe la production des cellules normales. Ceci conduirait à des défauts des cellules NK et ILCs, inhiberait la fonction anti-leucémique des cellules NK et rendrait les ILCs inefficaces, permettant ainsi le développement des maladies auto-immunes. Nous proposons de mettre à jours les mécanismes qui conduisent à la production de cellules NK et ILCs déficientes en étudiant leurs caractéristiques dans des échantillons de moelle osseuse et de sang de patients par comparaison à des donneurs sains. Ces travaux seront réalisés par des techniques de cytométrie en flux permettant de mesurer 18 paramètres simultanés sur les différentes cellules étudiées mais aussi par des techniques moléculaires (PCR quantitative) sur cellule unique. Des techniques in vitro de production de cellules NK et ILCs seront aussi mises au point à partir des cellules souches à l’origine des cellules sanguines. Ceci permettra d’évaluer les mécanismes médullaires de production des cellules NL et ILCs chez les malades. Ces expériences seront menées en présence ou non de molécules utilisées comme traitement des SMD (5-azacytidine). Les résultats permettront d’identifier les éléments à cibler pour restaurer une production NK et ILCs normale dans les SMD.
Valérie Ugo – Intérêt pronostic de la recherche de mutations additionnelles en NGS dans la myélofribrose
Les syndromes myéloprolifératifs sont des leucémies chroniques dont il existe plusieurs formes : la myélofibrose est une densification anormale de l’os concomitante de la prolifération des cellules leucémiques dans la moelle osseuse. La myélofibrose peut être présente dès le diagnostic de la maladie (on parle de myélofibrose primitive), ou survenir après plusieurs années d’évolution (myélofibrose secondaire). Actuellement, le diagnostic des syndromes myéloprolifératifs fait appel à la recherche de mutations acquises (= non héréditaires) touchant des gènes impliqués dans le développement de ces maladies, comme le gène JAK2, CARL ou MPL. Les techniques de séquençage de l’ADN de nouvelle génération (NGS) représentent un projet technologique important et permettent d’identifier de nombreuses autres mutations, dont le rôle dans la survenue de l’évolution de la myélofibrose est en pleine investigation. Ce projet vise à rechercher ces nouvelles mutations par NGS à partir d’échantillons prélevés chez environ 500 patients inclus dans l’observatoire national des myélofibroses du FIM, afin d’identifier des groupes de pronostic différent ce qui, à terme, aidera à définir la stratégie thérapeutique et à choisir des médicaments ciblés.
Appel à projet 2016
 Vahid Asnafi – Activation du TCR : nouvelle cible thérapeutique dans les LAL-T.
Vahid Asnafi – Activation du TCR : nouvelle cible thérapeutique dans les LAL-T.
Les leucémies aigües lymphoblastiques T (LAL-T) sont des cancers agressifs du sang. Les chances de survie, chez l’enfant et surtout chez l’adulte, y restent malheureusement encore limitées. Cela souligne la nécessité du développement de nouvelles approches thérapeutiques ciblant spécifiquement les cellules tumorales. Basés sur nos résultats publiées et préliminaires nous avons montré que : (i) la stimulation persistante du récepteur à l’antigène T (TC/CD3), exprimé par les LAL-T, a des propriétés anti-leucémiques chez la souris et chez l’homme (ii) l’immunothérapie par un anticorps ciblant la molécule TCR/CD3 permet d’améliorer la survie des souris de manière considérable. Nous souhaitons (a) approfondir et mieux explorer ce concept dans une série de LAL-T primaires, traités dans le protocole national GRAAL-2005 et évaluer davantage l’impact d’un traitement anti-CD2 dans des modèles précliniques ; (b) déchiffrer au niveau moléculaire les bases de la mort cellulaire observée. Nous établirons ainsi si cette approche peut être exploitée en clinique humaine pour améliorer la prise en change de ces cancers.

Alexandre Kauskot
Alexandre Kauskot – Thrombopénie/thrombopathie par désilyation des plaquettes dans la maladie de Willebrand de type 2B

Le facteur Willebrand (VWF) est une protéine clé de l’hémostase. Toute anomalie du VWF conduit à un syndrome hémorragique connu sous le nom de la maladie de Willebrand (von Willebrand disease, VWD) dont il existe plusieurs types. Parmi les anomalies constitutionnelles de l’hémostase, la VWD est classiquement la plus fréquente et la maladie de Willebrand de type 2B (VWD-type 2B) est probablement la plus complexe et est très hétérogène. La VWD-type 2B est caractérisée notamment par une thrombopénie (diminution de la quantité de plaquettes) inconstante, fluctuante, d’intensité variable. La thrombopénie peut avoir une origine centrale (moelle osseuse) et périphérique (circulation).
Nous souhaitons investiguer les causes moléculaires de la thrombopénie d’origine périphérique. Des modifications biochimiques apparaissent à la surface des plaquettes dans cette pathologie. Nous souhaitons dans ce projet comprendre comment ces modifications participent à la diminution de la quantité de plaquettes et s’il est possible de contrôler ce mécanisme en vue de restaurer un compte plaquettaire normal chez ces patients.

Fanny Drieux

Anaïs Pujals
Anaïs Pujals et Fanny Drieux – Caractérisation moléculaire des lymphomes T périphériques non-spécifiés : identification de sous-groupes moléculaires et impact clinique.
Les lymphomes T périphériques sont des cancers rares développés à partir de cellules immunitaires appelées lymphocytes T. En France, environ 1600 nouveaux cas sont diagnostiqués chaque année. Il s’agit de maladies très graves, les traitements chimiothérapiques actuellement disponibles étant peu efficaces (avec seulement 30 % de malades survivants à 5 ans).![]() Le diagnostic par examen au microscope est difficile, car il existe plusieurs maladies proches dont certaines sont encore mal définies. Notre projet a pour but de caractériser ces différents lymphomes T sur le plan génétique en utilisant différentes techniques de séquençage à haut débit.
Le diagnostic par examen au microscope est difficile, car il existe plusieurs maladies proches dont certaines sont encore mal définies. Notre projet a pour but de caractériser ces différents lymphomes T sur le plan génétique en utilisant différentes techniques de séquençage à haut débit.
Nous étudierons ensuite les conséquences biologiques de ces anomalies génétiques et leur conséquences cliniques notamment sur l’évolution des malades. Nous comptons identifier de nouveaux marqueurs permettant d’améliorer le diagnostic et le traitement des patients atteints.
Prix
Prix Brigitte Merand

DÉVELOPPER DES STRATÉGIES D’OPTIMISATION POUR AMÉLIORER LA RÉPONSE AUX THÉRAPIES DANS LES LEUCÉMIES AIGUËS MYELOÏDES (LAM)
Les chimiothérapies ou encore l’utilisation de certaines thérapies ciblées dans les leucémies aiguës myéloïdes (LAMs) sont efficaces pour éliminer rapidement une grande partie des cellules leucémiques, mais des rechutes sont malheureusement observées chez la plupart des patients, et se poursuivent fatalement par la résurgence de la maladie. Les projets que nous avons conduits jusqu’à présent consistent à identifier les leviers biologiques qui participent à la résistance des cellules tumorales à ces thérapies. Nous avons ainsi pu montrer que la reprogrammation métabolique des cellules leucémiques est un mécanisme biologique décisif pour promouvoir leur résistance aux thérapies de premières lignes ou ciblées. Enfin, en combinant des techniques de profilage et de criblages fonctionnels, nous avons identifié de nouveaux effecteurs fonctionnels – et défini leur mécanisme d’action in vitro et in vivo – afin de poser les jalons de schémas thérapeutiques optimisés.
Prix de thèse

Marie PASSET
AMÉLIORATION DU TRAITEMENT DES LEUCÉMIES AIGÜES MYÉLOÏDES (LAM)
Ce travail a permis d’étendre notre connaissance des leucémies aiguës lymphoblastiques B de l’adulte et de décrire deux nouvelles entités reconnues par la dernière classification consensus internationale. L’une définie par une mutation du gène PAX5, associée à un bon pronostic. La seconde qui touche les femmes jeunes est associée à un haut risque de rechute. La connaissance des anomalies en cause permet de dépister ces leucémies chez les patients dès le diagnostic. Ce travail ouvre la voie à de futures approches thérapeutiques de précision visant à identifier le meilleur traitement pour chaque patient.

Nicolas VALLET
MÉCANISMES BIOLOGIQUES IMPLIQUÉS DANS L’AUGMENTATION DU RISQUE DE RECHUTE DES HÉMOPATHIES MALIGNES PAR L’AZITHROMYCINE APRÈS ALLOGREFFE DE CELLULES SOUCHES HÉMATOPOÏÉTIQUES
L’allogreffe de cellules souches hématopoïétiques est un traitement curatif majeur des cancers du sang. Son effet dépend de l’activité antitumorale des lymphocytes T du donneur. Un antibiotique, l’azithromycine, peut compromettre son efficacité en inhibant directement l’activité antitumorale des lymphocytes T, et indirectement par une modification des réponses antitumorales médiée par l’altération du microbiote intestinal et de son métabolisme.
Prix Brigitte Merand

David MICHONNEAU
COMPRÉHENSION DES COMPLICATIONS IMMUNOLOGIQUES DE L’ALLOGREFFE DE CELLULES SOUCHES HÉMATOPOÏÉTIQUES
L’allogreffe de cellules souches hématopoïétique est un traitement qui permet d’obtenir une guérison de nombreuses hémopathies malignes et repose sur l’obtention d’une réponse anti-tumorale à partir des cellules immunitaires du donneur contre les cellules tumorales du patient (effet du greffon contre la tumeur, GVT).
J’ai consacré mon activité de recherche à la compréhension des mécanismes biologiques responsables des complications après une allogreffe de cellules souches hématopoïétiques.
Ces complications peuvent survenir lorsque le système immunitaire issu du greffon s’attaque aux patients (réaction du greffon contre l’hôte, ou GvHD) ou lorsque ce système immunitaire ne parvient pas à éliminer les cellules tumorales chez le patient (survenue d’une rechute post-allogreffe). La compréhension des mécanismes associés à la survenue de la GVHD et à la rechute est indispensable pour améliorer la prise en charge des patients allogreffés en réduisant les complications postgreffe.
Mes travaux ont eu pour but de mieux comprendre la façon dont le microbiote et son métabolisme interagissent avec le système immunitaire et contribue à la survenue de ces complications. Ainsi, ils ont d’abord permis de montrer que certaines bactéries composant le microbiote digestif sont capables de produire des composés chimiques, les dérivés indoles, à partir de notre alimentation. Ces molécules régulent la réponse immunitaire après une greffe et leur absence est associée avec l’apparition d’une GvHD aigue.
Par la suite, j’ai utilisé des approches dites de « multiomiques » pour décrire précisément la façon dont le système immunitaire est remodelé après la greffe. Ainsi, ces recherches ont permis de décrire les interactions complexes qui aboutissent à l’apparition d’une tolérance immunitaire après une allogreffe de moelle, c’est à dire un état au cours duquel les patients ne présentent ni GvHD, ni rechute, après l’arrêt des immunosuppresseurs.
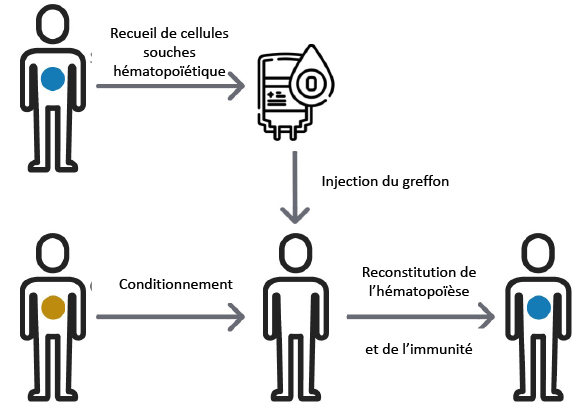
Parmi les mécanismes impliqués dans l’apparition de la tolérance, les modifications de la production des métabolites associées avec l’âge des receveurs était un des éléments clés qui participait à réguler la reconstitution du système immunitaire et à la balance entre un état de tolérance d’une part, ou la persistance d’une GvHD d’autre part. Enfin, mes travaux les plus récents se sont intéressés à la façon dont la prise d’un antibiotique courant, l’azithromycine, peut moduler la réponse immunitaire dirigée contre les cellules tumorales après la greffe. Ainsi, la prise chronique d’azithromycine est associée à une augmentation du risque de rechute des maladies du sang après une allogreffe, et ce risque est lié à l’action directe de l’azithromycine sur la réponse immunitaire. L’azithromycine est ainsi capable de modifier le fonctionnement des mitochondries, les usines énergétiques de la cellule, et de bloquer la capacité des lymphocytes à détruire des cellules cancéreuses.
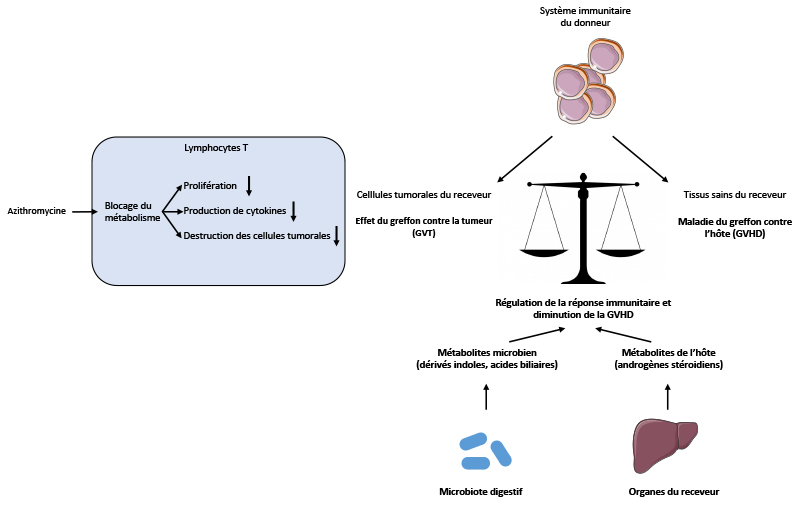
Ces résultats ont ainsi permis de mieux comprendre les voies biologiques qui sont nécessaires pour l’établissement d’une réponse anti-tumorale efficace, et dont la stimulation pourrait aider à diminuer le risque de rechute après la greffe et constituer de nouvelles pistes thérapeutiques.
Prix de thèse

Pierre Luc MOUCHEL
AMÉLIORATION DU TRAITEMENT DES LEUCÉMIES AIGÜES MYÉLOÏDES (LAM)
Mes travaux de thèses ont permis de montrer qu’une molécule naturelle issue du cholestérol, la Dendrogénine A, était capable d’augmenter l’effet des 2 principales chimiothérapies classiquement utilisées dans la lutte contre la leucémie aiguë myéloïde (LAM) : la cytarabine (ARAC) et l’idarubicine (IDA). Nous avons également pu montrer qu’une partie de la synergie entre l’IDA et l’ARAC passait par une inhibition des mitochondries des cellules de LAM par l’IDA. Ce nouveau mécanisme témoigne du rôle clef des mitochondries dans la LAM.

Vincent ALCAZER
CARACTÉRISATION DES POPULATIONS CELLULAIRES NORMALES ET TUMORALES ET DÉFINITION D’ÉPITOPES PARTAGÉS À PARTIR DES RÉTROVIRUS ENDOGÈNES HUMAINS (HERVSCARACTÉRISATION DES POPULATIONS CELLULAIRES NORMALES ET TUMORALES ET DÉFINITION D’ÉPITOPES PARTAGÉS À PARTIR DES RÉTROVIRUS ENDOGÈNES HUMAINS (HERVS)
Environ 8 % du génome humain est constitué de rétrovirus (HERVs). Silencieux à l’état sain, ces rétrovirus se réactivent fortement dans le cancer. Durant ma thèse, nous avons mis au point une méthode pour identifier des cibles à partir des HERVs exprimés dans n’importe quel cancer. L’application de cette méthode aux Leucémies Aiguës Myéloïdes nous a permis d’identifier des cibles thérapeutiques originales exprimées notamment par les cellules souches leucémiques.
Prix Brigitte Merand

Yacine BOULAFTALI
ÉTUDE DES SYSTÈMES DE RÉGULATION DE LA PLASMINE DANS LES NÉOPLASMES MYÉLOPROLIFÉRATIFS CONDUISANT À UNE MYÉLOFIBROSE PRIMAIRE ET DES PLAQUETTES DANS LA FIBROSE PULMOBAIRE
Les plaquettes sont garantes du maintien de l’intégrité vasculaire en empêchant les saignements. À côté de ces effets protecteurs, les plaquettes sont considérées comme proinflammatoires en interagissant avec les leucocytes contribuant ainsi aux maladies thromboinflammatoires. Les plaquettes et leurs précurseurs, les mégacaryocytes, peuvent réguler l’accumulation excessive de protéines matricielles comme lors de la (myelo)fibrose. Elles peuvent secréter des facteurs profibrosants et inhibiteurs de la plasmine, l’enzyme clé dans l’élimination du tissu fibreux. Notre projet portera sur l’étude (i) du système de régulation de la plasmine dans les néoplasmes myéloprolifératifs conduisant à une myélofibrose primaire et (ii) et des plaquettes dans la fibrose pulmonaire.
Prix de thèse

Damien LUQUE PAZ
PROFIL GÉNOMIQUES ET IMPACT PRONOSTIQUE DANS LES SYNDROMES MYÉLOPROLIFERATIFS
Les syndromes myéloprolifératifs sont des leucémies chroniques pouvant évoluer en des maladies plus sévères comme la myélofibrose ou la leucémie aiguë. Au cours de ce travail de thèse, nous avons recherché des mutations de l’ADN chez des patients atteints de syndromes myéloprolifératifs et avons montré l’intérêt de certaines de ces mutations afin d’identifier les patients à haut risque d’évolution.

Laurene FENWARTH
ÉTUDE DES ALTÉRATIONS GÉNOMIQUES DES LEUCÉMIES AIGUËS MYÉLOÏDES DE L’ADULTE : VERS UNE APPROCHE THÉRAPEUTIQUE PERSONNALISÉE ?
La leucémie aiguë myéloïde (LAM) correspond à un cancer du sang dont le pronostic demeure sombre avec un taux de guérison de 35-40 % chez l’adulte à l’heure actuelle. Le pronostic de cette maladie repose sur différents paramètres cliniques, biologiques et moléculaires. L’intégration de ces différents paramètres dans une approche multi-étapes permettrait de prédire de façon personnalisée le devenir de chaque patient et pourrait aider à optimiser sa prise en charge thérapeutique.
Prix Brigitte Merand

Alexandre Detappe
Alexandre Detappe – Travaux de développement de nanomatériaux pour améliorer le traitement du myélome multiple
Nos travaux de recherche se concentrent sur deux axes de développement de nanomatériaux pour améliorer le traitement du myélome multiple.
D’une part, nous développons des biomarqueurs d’imagerie IRM et TEP ciblant BCMA. Cette approche permet l’amélioration du diagnostic précoce du myélome multiple de manière plus efficace que leur concurrents, les traceurs immunoTEP.
D’autre part, nous développons de nouvelles approches thérapeutiques, avec entre autres un nouveau polymère permettant la délivrance contrôlée du triptique thérapeutique Revlimid (lenalidomide) / Velcade (bortezomib) / Dexamethasone) ou d’autres nouvelles molécules thérapeutiques (CELMod, PROTAC).
Enfin, suite à l’obtention d’une bourse ERC Starting Grant 2020, nous développons une nouvelle approche de bio ingénierie de cellules immunitaires à l’aide de la nanomédecine pour améliorer l’efficacité de l’immunothérapie.
Prix de thèses

Camille Roussel
Camille Roussel – Mise en évidence d’un marqueur sur les globules rouges permettant de prédire l’efficacité d’une transfusion
La forme de disque des globules rouges leur permet de se déformer de manière importante pour atteindre les petits vaisseaux sanguins et apporter l’oxygène dans tout l’organisme. Avant d’être transfusés, les globules rouges du donneur sont souvent conservés plusieurs semaines. Nous avons montré que certains globules rouges conservés deviennent plus sphériques et moins déformables. Cette dégradation entraine leur élimination rapide par la rate du receveur, rendant la transfusion moins efficace.

Hussein Issaoui
Hussein Issaoui – Le rôle de la région régulatrice en 3’ du locus des chaînes lourdes d’immunoglobulines dans le développement des lymphocytes B-1
Notre laboratoire s’intéresse à deux segments d’ADN qui contrôlent la production de nos anticorps. Notre objectif est de comprendre leurs rôles, leurs mécanismes de fonctionnement dans un contexte normal et l’effet qu’ils peuvent avoir lors de la formation d’un lymphome. A long terme, une meilleure compréhension des événements aboutissant au développement d’un lymphome, notamment par l’analyse de modèles animaux, est nécessaire et porteuse de promesses pour l’émergence de nouvelles perspectives thérapeutiques.
Prix Brigitte Merand

Pierre Sujobert
Pierre Sujobert – Mise en évidence de l’intérêt thérapeutique des activateurs d’AMPK dans les leucémies aiguës myéloïdes (LAM).
Mon activité de recherche vise à comprendre les mécanismes conduisant à la transformation maligne des cellules du sang, afin de découvrir de nouvelles pistes thérapeutiques pour les patients. Par exemple, nous avons montré que les cellules de leucémie aiguë myéloïde étaient particulièrement sensibles aux agents activant la protéine AMPK, ce qui ouvre des perspectives thérapeutiques originales. Je cherche désormais à caractériser les fonctions cellulaires de deux gènes fréquemment altérés dans les lymphomes, afin de trouver des approches thérapeutiques personnalisées pour les patients ayant ces altérations. En parallèle, j’effectue des travaux plus proches de la réalité clinique, dont l’objectif est de comprendre comment utiliser au mieux l’information issue de l’analyse génétique des hémopathies pour améliorer la prise en charge des patients.
Prix de thèses

Bérangère Joly
Bérangère Joly – Déficit fonctionnel sévère en ADAMTS13 (protéase spécifique de clivage du facteur Willebrand) : physiopathologie du purpura thrombotique thrombocytopénique de l’enfant.
Le purpura thrombotique thrombocytopénique (PTT) à révélation pédiatrique est une maladie très rare, définie par un déficit sévère en ADAMTS13. Ce déficit peut être héréditaire (mutation du gène) ou acquis (anticorps dirigés contre ADAMTS13). La cohorte pédiatrique française de PTT a été entièrement caractérisée sur les plans épidémiologique, phénotypique et génotypique. La mise au point d’outils innovants a permis d’explorer les changements de conformation d’ADAMTS13 associés aux mutations du gène ou aux anticorps anti-ADAMTS13.

Aurore Touzart
Aurore Touzart – Leucémies Aiguës Lymphoblastiques T (LAL-T) et dérégulation épigénétique
Dans mon travail de thèse, je me suis intéressée à un type particulier de leucémie aiguë, la leucémie aiguë lymphoblastique T, qui peut toucher l´enfant et l´adulte et dont le pronostic reste médiocre. J´ai cherché à comprendre comment des mécanismes de dérégulation épigénétique pouvaient intervenir dans ces leucémies et dans la réponse des patients au traitement. L´épigénétique regroupe des phénomènes qui participent à la régulation de l’expression du génome mais sans modifications irréversibles de ce dernier.
Prix Brigitte Merand

Isabelle Plo-Azevedo
Isabelle Plo-Azevedo
Les néoplasmes myéloprolifératifs (NMP) classiques sont des maladies hématologiques clonales et malignes qui regroupent la thrombocytémie essentielle, la polyglobulie de Vaquez et la myélofibrose qui est la plus grave. Ils peuvent évoluer à plus ou moins long terme vers des leucémies aiguës secondaires souvent incurables. Ils sont dus à des aberrations génétiques qui inhibent le contrôle physiologique de la prolifération ce qui résulte dans une expansion incontrôlée de la production des globules rouges, des plaquettes ou des cellules granuleuses. Notre objectif est d’étudier l’étiologie et les mécanismes physiopathologiques des NMP pour développer des thérapies ciblées préventives et curatives.

Salomon Manier
Salomon Manier
Le myélome multiple (MM) est une hémopathie maligne incurable. Les anomalies du gène MYC sont responsables de la progression de la maladie. Cependant, il n’existe pas de traitement permettant de cibler MYC directement. Dans ce travail, nous avons identifié que les cellules surexprimant MYC nécessitent une forte activité de synthèse des protéines et qu’une nouvelle classe de médicaments appelée Rocaglate inhibe la synthèse des protéines et détruit ainsi préférentiellement les cellules de MM surexprimant MYC.

Nicolas Duployez
Nicolas Duployez – Étude des altérations génomiques acquises dans les leucémies aiguës myéloïdes impliquant le core binding factor
Les gènes RUNX1 et CBFB codent pour les sous-unités du core binding factor (CBF), facteur de transcription hétérodimérique essentiel de l’hématopoïèse définitive. La dérégulation du CBF est l’une des anomalies les plus fréquemment rencontrées dans les hémopathies malignes. Puisque la perturbation seule du CBF est insuffisante au développement d’une leucémie aiguë myéloïde (LAM), les LAM impliquant le CBF sont considérées comme des modèles de leucémogénèse multi-étapes, nécessitant la coopération d’anomalies génétiques additionnelles. Dans ce travail, je me suis intéressé aux LAM de type CBF, caractérisées soit par une t(8;21)/fusion RUNX1-RUNX1T1 soit par une inv(16)/fusion CBFB-MYH11, ainsi qu’aux LAM avec mutations germinales de RUNX1 (définissant la thrombopénie familiale avec prédisposition aux leucémies aiguës ou FPD/AML). Afin d’identifier des anomalies additionnelles, nous avons étudié les prélèvements de patients atteints de LAM CBF inclus dans les essais français ELAM02 (0-18 ans) et CBF2006 (18-60 ans) par séquençage à haut débit (n=215) et single nucléotide polymorphism-array (n=198). Les échantillons de 25 individus atteints de FPD/AML (issus de 15 familles), diagnostiqués entre 2005 et 2014, ont également été séquencés au stade thrombopénique et au moment de la transformation en leucémie aiguë. Dans les LAM CBF, les mutations activatrices des voies tyrosines kinases (TK) sont les événements les plus fréquents quel que soit le sous-type de LAM CBF, comme cela a déjà été rapporté dans d’autres études. En revanche, les mutations affectant les gènes du remodelage chromatinien (notamment ASXL1 et son homologue ASXL2) ou du complexe de la cohésine sont identifiées à des fréquences élevées (41% et 18% respectivement) dans les LAM avec t(8;21) tandis qu’elles sont pratiquement absentes dans les LAM avec inv(16). Dans les LAM avec t(8;21), la coexistence de ces mutations avec les mutations de type TK est associée à un pronostic défavorable suggérant une synergie entre ces événements. D’autres événements fréquemment retrouvés incluent les mutations de ZBTB7A et DHX15 dans les LAM avec t(8;21) (20% et 6% respectivement) et les délétions/mutations de FOXP1 dans les LAM avec inv(16) (7%). Enfin, nous avons décrit la perturbation de CCDC26 comme une possible lésion associée à une signalisation aberrante des TK dans les LAM CBF (4,5% des cas). Dans les FPD/AML, l’analyse mutationnelle a révélé l’acquisition d’un deuxième événement impliquant RUNX1 chez tous les patients ayant développé une LAM. Ce deuxième événement correspondait soit à une mutation somatique du second allèle de RUNX1 soit à la duplication de la mutation germinale de RUNX1 (par perte d’hétérozygotie sans anomalie du nombre de copies ou trisomie 21 acquise). En pratique clinique, cela suggère que la présence de deux mutations différentes de RUNX1 ou d’une seule mutation avec un ratio allélique supérieur à 50% chez un patient atteint de LAM doit alerter sur la possibilité d’un syndrome FPD/AML sous-jacent.
Prix Brigitte Merand

Thomas Mercher
Mutation de l’ADN dans la la leucémie infantile
Thomas Mercher a obtenu une thèse de génétique humaine (Université Pierre et Marie Curie, Paris) en 2003, réalisé un post-doctorat au Brigham & Women’s Hospital (Harvard Medical School, Boston, USA) puis intégré l’Inserm en 2008. Il est actuellement directeur de recherche et responsable d’équipe de l’Institut Gustave Roussy (Villejuif). L’équipe qu’il anime a pour objectif d’identifier les mutations de l’ADN observées dans les leucémies de l’enfant, de comprendre leurs mécanismes de transformation en développant des modèles d’études physiologiques. A terme, l’objectif est d’identifier de nouvelles stratégies thérapeutiques. L’équipe a concentré ses analyses sur les leucémies aiguës mégacaryoblastiques (LAM-M7) de l’enfant qui sont associées à un mauvais pronostic et a récemment mis en évidence de nouveaux mécanismes d’action d’une protéine de fusion anormale.
Prix de thèses

Alexis Saintamand
Alexis Saintamand a soutenu sa thèse sur la régulation génétique du développement des lymphocytes B au sein de l’UMR CNRS 7276 de Limoges en 2016. Il travaille désormais dans l’U1236 Inserm à Rennes sur les mécanismes à l’origine du développement du lymphome folliculaire.
Les lymphocytes B sont des cellules du système immunitaire chargées notamment de produire des anticorps (ou immunoglobulines). Les anticorps sont des molécules complexes capables de reconnaitre les éléments pathogènes pour aider à leur élimination. Chaque anticorps ne pouvant reconnaitre qu’une seule molécule, l’organisme doit en générer une grande diversité. Pour cela, les lymphocytes B vont devoir remanier une partie de leur génome par une succession de cassures, délétions, réparations et mutations. Phénomènes dangereux, potentiellement cancérogène, ces mécanismes sont étroitement régulés par des séquences particulières d’ADN réparties tout au long du segment de chromosome qui code les anticorps. Alexis Saintamand a travaillé en particulier sur l’une de ces régions, nommée « région régulatrice localisée en 3’ du locus IgH » (ou 3’RR), connue pour réguler la synthèse des anticorps. Outre ce rôle, la 3’RR possède également un côté délétère. En effet, les phénomènes de cassures et mutations se produisant dans l’ADN des lymphocytes B font du locus IgH une zone critique de translocations, c’est-à-dire d’échange de fragments entre les chromosomes. Lorsqu’un gène est déplacé au locus IgH, son expression est activée par la 3’RR. Or, certains gènes, lorsqu’ils sont dérégulés, rendent la cellule cancéreuse.

Angèle Gros
Angèle Gros a réalisé sa thèse intitulée « Les plaquettes dans les réactions inflammatoires : maintien de l’intégrité vasculaire et régulation des neutrophiles » dans l’unité Inserm 1148 sous la direction du Docteur Martine Jandrot-Perrus et du Docteur Benoît Ho-Tin-Noe.
Les plaquettes assurent le maintien de l’intégrité vasculaire et l’arrêt des saignements en cas de blessures. En plus de ce rôle dans l’hémostase primaire, les plaquettes participent à la réponse immunitaire. Le travail de cette thèse s’est intéressé au rôle des plaquettes dans les réactions inflammatoires. Angèle Gros a cherché à comprendre comment les plaquettes prévenaient la survenue des saignements inflammatoires et à caractériser la régulation des neutrophiles par les plaquettes. Ce travail constitue une première étape vers de nouvelles applications thérapeutiques des médicaments antiplaquettaires.
Prix Jeune chercheur «Brigitte MÉRAND» 2016

Benoit HO-TIN-NOE et Pierre MICHEL
Le prix du jeune chercheur « Brigitte Merand » 2016 a été attribué à Benoît Ho-Tin-Noé. Benoît Ho-Tin-Noé est un jeune scientifique âgé de 37 ans. Il a fait ses études de Sciences à l’Université 7 Denis Diderot, un post-doctorat de 2 ans à Harvard Medical School, Boston, USA et a été recruté en 2009 à l’Inserm comme chargé de recherche. De 2009 à 2013 il a travaillé dans l’unité Inserm U698, sous la direction de Jean-Baptiste Michel, puis en 2004, il a intégré le laboratoire de Didier Letourneur, Inserm U1148, à l’hôpital Bichat.
Son thème principal de recherche s’inscrit dans la continuité de ses travaux de post-doctorat sur le rôle vasculo-protecteur exercé par les plaquettes dans différentes situations inflammatoires et tumorales. Ses travaux étudient la régulation par les plaquettes des activités des cellules immunitaires et notamment des polynucléaires neutrophiles. Benoît Ho-Tin-Noé cherche à comprendre les mécanismes à l’origine des effets protecteurs des plaquettes vis-à-vis des vaisseaux tumoraux. Son but est d’explorer les possibilités d’interférer pharmacologiquement avec cette action pro-tumorale des plaquettes de façon à l’inhiber et faciliter ainsi l’effet des chimiothérapies. Benoît Ho-Tin-Noé est déjà auteur et co-auteur de 35 publications internationales, est régulièrement invité à présenter ses travaux dans des congrès en France et à l’étranger Il a participé à divers jurys de thèse et a aussi obtenu cette année des contrats de recherche permettant des financements importants pour ses travaux. Benoît Ho-Tin-Noé est également éditeur principal dans le journal « Plaelets » ce qui témoigne de sa reconnaissance internationale.
Prix de thèses

Amélie TRINQUAND et Hubert LEVY-LAMBERT
Le prix de thèse à été attribué à Amélie Trinquand qui est médecin biologiste et assistante hospitalo-universitaire dans le laboratoire d’hématologie de l’hôpital Necker-Enfants-Malades. Durant le cursus de ses études, après l’interna Amélie Trinquand a fait un Master 2 puis une thèse d’Université de 2012 à 2015 sous la direction du Professeur Vahid Asnafi, dans le laboratoire Inserm U1151 à l’hôpital Necker. La soutenance de sa thèse a eu lieu le 23 octobre 2015. Le sujet de son travail de recherche était « Signalisation TCR et leucémies aigues lymphoblastiques T ».
Les leucémies aigues lymphoblastiques T (LAL-T) sont des hémopathies malignes rares causées par la prolifération de cellules immatures T (dérivées des lymphocytes thymiques T) bloquées à un stade donné de leur différenciation. Leur oncogenèse est multigénique ; ainsi plusieurs anomalies chez le même patient sont à l’origine du blocage de différenciation cellulaire et de gains de fonctions de type prolifération cellulaire et résistante à l’apoptose.
Son travail de thèse a consisté à analyser si l’activation et la signalisation du récepteur des lymphocytes T (TCR) pouvaient être impliquées dans la biologie de cette hémopathie. Elle a montré que l’induction d’une signalisation persistante/forte du TCR provoque la mort massive des LAL-T primaires TCR + et exerce une fonction anti leucémique in vivo, fournissant un fort rationnel en faveur d’une thérapie ciblée, basée sur l’activation du TCR. Elle a également étudié la fréquence et l’impact pronostique des anomalies de la signalisation du TCR dans une série de LAL-T de l’adulte. Enfin, elle a mis en évidence une anomalie d’une kinase en aval du TCR qui pourrait être une cible thérapeutique dans les LAL-T. Au total, les travaux de la thèse d’Amélie Trinquand montrent que la signalisation du TRC et son activation sont impliquées dans la biologie des LAL-T. La qualité de son travail de recherche a été reconnue par la publication de 3 articles qu’elle signe en premier auteur dans d’excellents journaux internationaux dont Cancer Discovery et Journal of Clinical Oncology.
Premier Prix Brigitte Merand
Le 18 mars 2018, à l’Hôtel des Arts et Métiers à Paris, en présence des Professeurs Jean-Jacques SOTTO, Président de Force Hémato, Hélène MERLE-BERAL, François DREYFUS et Philippe COLOMBAT, le premier prix créé par le Fonds de recherche clinique en hématologie pour récompenser et soutenir les travaux d’un chercheur a été remis au Professeur Florence N’GUYEN KHAC de la Pitié Salpêtrière.
Ce prix qui sera attribué chaque année, portera le nom de Brigitte MERAND en hommage à une malade qui a lutté avec un grand courage contre sa maladie.

Florence Nguyen Khac
Florence Nguyen-Khac a reçu le prix Brigitte Merand pour l’ensemble de ses travaux publiés en 2014 sur la LLC : trois études qu’elle a dirigées et deux collaborations. Elle a cherché à comprendre les conséquences fonctionnelles des nombreuses anomalies génomiques observées dans la LLC, qui peuvent jouer un rôle dans le développement, la progression de la maladie et la résistance au traitement.
La leucémie lymphoïde chronique (LLC) est la leucémie la plus fréquente de l’adulte dans les pays caucasiens, et se caractérise par une prolifération et une accumulation de lymphocytes B matures dans le sang, la moelle osseuse, les ganglions et la rate. Il s’agit d’une maladie classiquement indolente pendant de nombreuses années, avec cependant une grande variabilité d’évolution. La LLC est encore incurable aujourd’hui, avec des rechutes inévitables, et l’apparition de résistances aux drogues classiques utilisées. Les évènements oncogéniques à l’origine de la LLC sont à ce jour peu connus. La caractérisation cytogénétique et moléculaire des cellules de LLC est primordiale pour avancer dans la compréhension et le traitement de cette leucémie.
Projets de Recherche Infirmier

Thomas BERT
L’HYPNOSE : UN CHEMIN VERS L’APAISEMENT
En hématologie, la majorité des patients sont traités pour un lymphome, un myélome ou une leucémie. Leur parcours de soins est souvent long et éprouvant, ponctué de différentes cures de chimiothérapie, d’immunothérapie ou d’auto/allogreffe. Ils sont régulièrement confrontés à des douleurs chroniques, à un épuisement ou à de l’anxiété. Les traitements utilisés sont également pourvoyeurs d’effets secondaires (nausées, fatigues, …). La médecine classique semble parfois être démunie et inadaptée pour répondre à ces problématiques. L’hypno-analgésie est une technique complémentaire dans la prise en charge de ces maux. Elle consiste à amener le patient à moduler ses ressentis négatifs en focalisant son attention mentale sur autre chose. Cet outil est de plus en plus utilisé comme soin de support à l’hôpital. En hématologie à l’hôpital Lyon-Sud, un accompagnement en hypnose est proposé aux patients dans le service et en consultation ambulatoire. Dans la littérature scientifique, des études ont montré que l’hypnose permet de réduire le stress des patientes traitées pour un cancer du sein ou de diminuer les douleurs lors des gestes invasifs. Le but du travail de recherche infirmier « L’hypnose : un chemin vers l’apaisement » est de mettre en évidence l’impact positif de l’hypno-analgésie sur l’anxiété et la qualité de vie des patients en hématologie, en comparant un groupe de patients bénéficiant de séances d’hypnose et un groupe contrôle. Cette étude sera menée à partir d’avril 2023 dans le service d’hématologie du Centre Hospitalier Lyon-Sud.
Alizée Soldati – APALLO : activité physique adaptée post allogreffe de cellules souches hématopoïétiques

Alizée Soldati
Les patients atteints de maladies hématologiques malignes reçoivent des traitements lourds et longs. Parmi ces traitements, il y a l’allogreffe de cellules souches hématopoïétiques. Cette procédure implique une hospitalisation de 5 à 6 semaines avec un suivi hebdomadaire durant plusieurs mois à la suite de celle-ci et un risque important de complications durant l’hospitalisation et durant les mois qui suivent. L’objectif de cette procédure est d’obtenir une rémission complète de la maladie sans rechute en remplaçant les cellules de la moelle osseuse du patient par celles d’un donneur sain. Les patients sont affaiblis, ont des défenses immunitaires immatures et prennent de nombreux traitements. Ils ont parfois des difficultés pour réaliser les gestes de la vie quotidienne, s’alimenter correctement, et mettent plusieurs mois avant de retrouver une vie dite « normale » comme avant le diagnostic.
Des études ont montré le grand intérêt de la pratique d’une activité physique durant et après le traitement contre le cancer dans certaines spécialités comme le cancer colorectal et ainsi se sont développés des centres et associations proposant des séances collectives pour tous les patients atteints de cancer. Malheureusement, les patients allogreffés ne peuvent pas participer à des activités collectives durant les premiers mois après l’hospitalisation du fait de leur système immunitaire immature et sont souvent trop fatigués pour se déplacer.
Cette étude a pour objectif de définir l’impact de l’activité physique adaptée au patient et individuelle sur la récupération après allogreffe. L’objectif secondaire est de permettre l’accès à cette activité physique à tous les patients afin d’améliorer la qualité de vie après les traitements pour le cancer et de diminuer les complications liées à la fatigue, à la dénutrition et à la perte musculaire.
Le projet sera réalisé grâce à une collaboration entre le service d’hématologie et la société EXPERF, spécialisée en soins et perfusions à domicile. La coordinatrice des soins de la société apportera son aide sur l’organisation des séances d’activité physique adaptée entre l’enseignant et les patients. Elle réalisera également un compte-rendu de chaque atelier qui permettra de faire le lien entre les séances au domicile et le suivi hospitalier.
Marie-Pierre Dann – Epreuve(s) de Damoclès : Sciences Humaines et Sociales, démarche d’accompagnement dans l’annonce de la leucémie lymphoïde chronique, qualité de vie

Damn Marie-Pierre
La leucémie lymphoïde chronique (LLC) touche quatre à cinq mille personnes par an en France. Parmi elles, deux tiers n’ont pas de symptômes, et la moitié de ces derniers n’aura jamais ni de symptômes, ni de traitement. Le suivi de ces personnes “malades mais en bonne santé” est restreint, et consiste généralement en une visite de contrôle tous les 6 à 12 mois. Le projet Damoclès vise à mieux comprendre quels sont les besoins, questions et angoisses de ces patients diagnostiqués avec une maladie parfois grave et incurable alors qu’ils n’ont ni symptômes, ni besoin de traitement. Le projet Damoclès rassemble une équipe interdisciplinaire d’infirmiers, de psychologues et de philosophes autour de l’élaboration et de la mise en circulation d’un questionnaire auprès de deux cohortes de patients (une cohorte contrôle qui n’est pas traitée, et une autre cohorte déjà incluse dans un essai thérapeutique, traitée à titre préemptif). Ce questionnaire permettra d’obtenir des données qualitatives et quantitatives sur le vécu et les besoins de ces patients, ainsi que de mener une étude comparative entre ces deux cohortes afin de mieux comprendre en quoi et comment le fait de proposer une forme de traitement dès les phases les plus précoces de la maladie peut impacter sur le vécu des patients. Il s’agit donc d’examiner et de comprendre les expériences subjectives du patient après l’annonce d’une LLC avec (ou non) abstention thérapeutique. Les résultats ainsi obtenus permettront d’identifier quelles formes d’accompagnement il est possible et nécessaire de proposer à ces personnes.